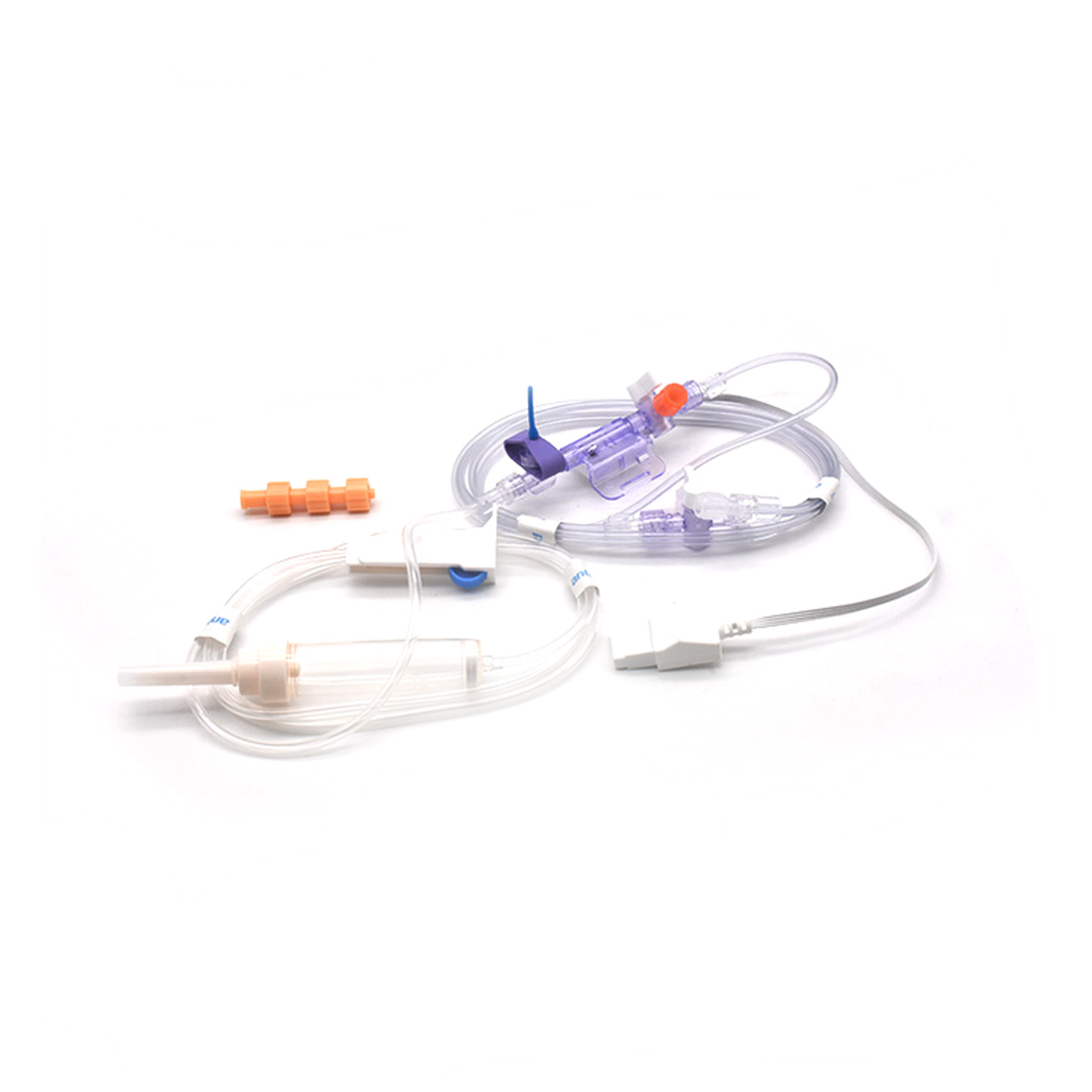
Hiểu về Cảm biến IBP và Ý nghĩa Lâm sàng của Chúng
Bộ chuyển đổi IBP là gì và nó hoạt động như thế nào?
Các bộ chuyển đổi đo huyết áp xâm lấn (IBP) là những thiết bị y tế thực hiện việc ghi nhận các chỉ số huyết áp trực tiếp từ động mạch thông qua ống thông, sau đó chuyển đổi thành tín hiệu điện để bác sĩ theo dõi tình trạng trong thời gian thực. Những thiết bị này thường được trang bị cảm biến vô trùng nhằm phát hiện cả những thay đổi nhỏ nhất về mức độ huyết áp, rồi truyền toàn bộ dữ liệu này đến máy theo dõi thông qua hệ thống chứa đầy dịch. Các mẫu thiết bị mới hiện nay đạt độ chính xác khoảng cộng trừ 2 mmHg, điều này rất quan trọng khi cần phát hiện kịp thời các tình trạng tụt huyết áp hay tăng huyết áp nguy hiểm trong quá trình phẫu thuật. Một nghiên cứu công bố năm ngoái trên Tạp chí Y học Chăm sóc Đặc biệt cho thấy một điểm thú vị – bệnh nhân được theo dõi bằng các bộ chuyển đổi chính xác này đã được phát hiện các cơn huyết áp thấp sớm hơn 42% so với các kỹ thuật không xâm lấn thông thường. Loại cảnh báo sớm như vậy tạo ra sự khác biệt lớn trong phòng mổ, nơi mà từng giây đều mang tính sống còn.
Ý nghĩa lâm sàng của việc theo dõi huyết áp xâm lấn chính xác
Đối với những bệnh nhân cần dùng thuốc vận mạch hoặc hỗ trợ ổn định tuần hoàn, việc theo dõi huyết áp xâm lấn (IBP) vẫn được coi là phương pháp tốt nhất. Các phương pháp dao động kế không đáp ứng được khi chúng ta cần những chỉ số theo từng nhịp tim, vốn rất quan trọng trong các trường hợp sốc nhiễm trùng, sau tai nạn nghiêm trọng hoặc trong quá trình phẫu thuật tim. Các bác sĩ dựa vào tốc độ phản hồi nhanh của các thiết bị này, thường dưới một giây, để đưa ra quyết định về các biện pháp như truyền dịch. Kinh nghiệm lâm sàng cho thấy rằng ngay cả việc trì hoãn năm phút để điều chỉnh các vấn đề huyết áp cũng có thể dẫn đến tỷ lệ tử vong tăng khoảng 15%. Một khảo sát gần đây về thực hành tại các đơn vị chăm sóc tích cực cũng cho thấy điều thú vị: các bệnh viện áp dụng thiết lập chuẩn cho cảm biến IBP đã ghi nhận sai sót khi điều chỉnh thuốc giảm khoảng 31% tại các khoa chăm sóc tích cực.
Chứng nhận CE và Tuân thủ EU MDR đối với Cảm biến IBP
Chứng nhận CE nghĩa là gì đối với an toàn và hiệu suất của thiết bị y tế
Việc đạt chứng nhận CE theo Quy định Thiết bị Y tế EU (MDR) về cơ bản có nghĩa là bộ chuyển đổi IBP đã vượt qua các kiểm tra an toàn nghiêm ngặt cần thiết để bán tại châu Âu. Dấu CE cho thấy sản phẩm tuân thủ các tiêu chuẩn quan trọng như IEC 60601 đối với thiết bị điện tử y tế và đồng thời đáp ứng các quy định quản lý rủi ro theo ISO 14971. Đối với các thiết bị tuân thủ quy định MDR, có một quy trình toàn diện nhằm kiểm tra mức độ tương thích an toàn của các vật liệu tiếp xúc với bệnh nhân, đảm bảo rằng chúng sẽ không gây tổn thương tế bào hay kích ứng. Hầu hết các thiết bị theo dõi huyết áp thuộc nhóm phân loại IIa hoặc cao hơn, điều này có nghĩa là nhà sản xuất không thể tự công bố sự phù hợp mà cần các chuyên gia độc lập xác minh mọi thứ hoạt động đúng trước khi đưa sản phẩm ra thị trường.
Các bước để đạt dấu CE theo MDR EU đối với bộ chuyển đổi IBP
- Phân loại thiết bị : Xác định phân loại rủi ro (thông thường là nhóm IIa đối với bộ chuyển đổi IBP) theo Phụ lục VIII của MDR
- Thiết lập hệ thống quản lý chất lượng (QMS) : Thiết lập hệ thống quản lý chất lượng được chứng nhận ISO 13485.
- Chuẩn bị tài liệu kỹ thuật : Tổng hợp các thông số thiết kế, đánh giá rủi ro và báo cáo đánh giá lâm sàng.
- Tìm kiếm Tổ chức được Chỉ định : Đối với thiết bị loại IIa trở lên, thực hiện kiểm toán các hồ sơ kỹ thuật và quy trình sản xuất.
- Dán nhãn CE : Sau đánh giá sự phù hợp, đăng ký thiết bị vào EUDAMED. Theo SimplerQMS, 89% sự chậm trễ xuất phát từ việc đánh giá lâm sàng không đầy đủ hoặc tài liệu QMS đã lỗi thời.
Vai trò của Tổ chức được Chỉ định trong việc xác nhận sự tuân thủ CE
Các Tổ chức được chỉ định, hay còn gọi tắt là NB, đóng vai trò là các bên kiểm toán độc lập đối với thiết bị y tế thuộc nhóm có nguy cơ cao hơn. Công việc của họ bao gồm rà soát toàn bộ tài liệu kỹ thuật và thực tế đến tận các cơ sở sản xuất để tiến hành kiểm tra. Khi xét riêng các cảm biến IBP, các tổ chức này sẽ kiểm tra xem mọi thứ có tuân thủ các yêu cầu nêu trong Phụ lục II của Quy định Thiết bị Y tế (MDR) hay không. Điều này bao gồm việc đảm bảo rằng các phiên bản kỹ thuật số phải có xác nhận phần mềm đầy đủ và các sản phẩm dùng một lần phải đáp ứng các tiêu chuẩn vô trùng nghiêm ngặt. Theo một khảo sát gần đây năm 2023, hầu hết các vấn đề mà các NB phát hiện đều bắt nguồn từ hai lĩnh vực chính: các công ty chưa xây dựng chiến lược giám sát sau khi lưu hành thị trường một cách vững chắc, hoặc không cung cấp đủ dữ liệu về tính tương thích sinh học. Những phát hiện này làm nổi bật những điểm mà các nhà sản xuất cần tập trung cải thiện nếu muốn vượt qua các cuộc kiểm toán một cách thành công.
Tự chứng nhận so với Đánh giá bởi Bên thứ ba đối với thiết bị Loại IIa
Trong khi các thiết bị không vô trùng loại I có thể tự chứng nhận, các cảm biến IBP loại IIa yêu cầu sự giám sát bắt buộc từ Tổ chức được chỉ định. Việc tự chứng nhận chỉ áp dụng cho các phụ kiện ít rủi ro như cáp tái sử dụng; các thành phần cảm biến áp lực phải trải qua đánh giá đầy đủ bởi bên thứ ba. Các cập nhật gần đây của Quy định Thiết bị y tế (MDR) yêu cầu kiểm toán hàng năm bởi Tổ chức được chỉ định đối với các nhà sản xuất thiết bị loại IIa, làm giảm 34% số trường hợp lưu hành trái phép kể từ năm 2021.
Tiêu chuẩn Sinh tương thích ISO 10993 và Tiêu chuẩn Quản lý Rủi ro ISO 14971
Tại sao tiêu chuẩn ISO 10993 lại quan trọng đối với các thành phần tiếp xúc bệnh nhân trong hệ thống IBP
Đối với các cảm biến IBP tiếp xúc trực tiếp với bệnh nhân, việc tuân thủ các tiêu chuẩn ISO 10993 gần như là bắt buộc nếu chúng ta muốn tránh những phản ứng sinh học đáng ghét. Mục đích chính của tiêu chuẩn này là xác định điều gì xảy ra khi con người tiếp xúc trong thời gian dài với các loại vật liệu như bộ phận nhựa, keo dán dùng trong lắp ráp và các chi tiết cảm biến khác nhau. Theo nghiên cứu được công bố năm ngoái, khoảng một trên năm trường hợp kích ứng da được báo cáo tại các bệnh viện xuất phát từ các thiết bị y tế được làm bằng vật liệu không đáp ứng yêu cầu ISO 10993-10. Các công ty tích hợp những yêu cầu này vào quy trình sản xuất thông qua Kế hoạch Đánh giá Sinh học phù hợp thường gặp ít vấn đề hơn vì họ kiểm tra cách các vật liệu khác nhau phản ứng khi tiếp xúc với dịch cơ thể và mô thật sự theo thời gian.
Đánh giá nguy cơ độc tế bào, dị ứng và kích ứng
ISO 10993 yêu cầu ba bài kiểm tra cốt lõi đối với vật liệu cảm biến IBP:
- Độc tính tế bào (ISO 10993-5): Đo lường khả năng sống sót của tế bào sau khi tiếp xúc với vật liệu (cho phép tối đa 70% tế bào chết)
- Gây nhạy cảm (ISO 10993-10): Đánh giá tiềm năng phản ứng dị ứng bằng các xét nghiệm tăng cường trên chuột lang
- Kích ứng (ISO 10993-10): Đánh giá nguy cơ viêm tại chỗ thông qua thử nghiệm dán trên biểu bì
Các thiết bị không đạt bất kỳ thử nghiệm nào có tỷ lệ thu hồi của FDA cao hơn 89% so với các lựa chọn tuân thủ đầy đủ (Báo cáo Thiết bị Y tế, 2022).
Nghiên cứu điển hình: Những thất bại trong lựa chọn vật liệu dẫn đến vi phạm quy định
Một cuộc kiểm toán EU MDR năm 2021 cho thấy 32% bộ chuyển đổi IBP bị từ chối sử dụng ống silicone không có chứng nhận ISO 10993-33 cho tiếp xúc máu kéo dài. Việc thu hồi 2,7 triệu USD của nhà sản xuất xuất phát từ:
- Giả định rằng vật liệu "dùng trong y tế" tự động đáp ứng các tiêu chuẩn tương thích sinh học
- Bỏ qua các thử nghiệm lão hóa nhân tạo để đánh giá rủi ro thôi nhiễm hóa chất
- Tài liệu không đầy đủ về tác động của tiệt trùng đến độ an toàn của vật liệu
Tích hợp Kiểm tra Tương thích sinh học với Quản lý rủi ro theo ISO 14971
Kết hợp dữ liệu ISO 10993 với khung quản lý rủi ro theo ISO 14971 cho phép các nhà sản xuất:
- Định lượng các rủi ro sinh học bằng ma trận mức độ nghiêm trọng/xác suất mối nguy
- Thực hiện các biện pháp kiểm soát như thay thế vật liệu hoặc phủ lớp vật liệu tương thích sinh học
- Giám sát các sự kiện bất lợi sau khi lưu hành thông qua các quy trình phù hợp với ISO 13485
Cách tiếp cận kép này làm giảm 41% số thay đổi thiết kế ở giai đoạn cuối so với việc chỉ thực hiện kiểm tra tương thích sinh học đơn lẻ (Tiêu chuẩn chất lượng MedTech, 2023).
Phê duyệt FDA và Các yêu cầu quy định toàn cầu đối với cảm biến IBP
Quy trình phê duyệt FDA 510(k) và Tính tương đương đáng kể
Đối với các nhà sản xuất thiết bị y tế, việc được thông qua chương trình 510(k) của FDA đồng nghĩa với việc chứng minh sản phẩm của họ về cơ bản tương đương với một sản phẩm đã có trên thị trường. Theo số liệu ngành, khoảng ba phần tư thiết bị đã được phê duyệt theo cách này vào năm 2023. Việc đáp ứng các yêu cầu này đòi hỏi khối lượng công việc thử nghiệm nghiêm ngặt và tuân thủ chặt chẽ các quy định tại Phần 820 CFR 21 về hệ thống chất lượng. Các công ty đã có chứng nhận ISO 13485 thường thấy hồ sơ của họ được FDA xử lý nhanh hơn khoảng 30 phần trăm. Điều này dường như bắt nguồn từ việc hồ sơ và tài liệu được tổ chức tốt hơn, phù hợp với những gì cơ quan quản lý mong đợi trong quá trình thẩm định.
Thách thức hài hòa: Sự khác biệt giữa EU MDR, FDA và các thị trường khác
Các yêu cầu khu vực khác nhau tạo ra sự phức tạp trong tuân thủ:
- EU MDR yêu cầu đánh giá lâm sàng để được dán nhãn CE
- FDA nhấn mạnh sự tương đương dựa trên sản phẩm tiền đề
- PMDA của Nhật Bản yêu cầu tuân thủ ISO 80369-6 đối với các kết nối tủy sống
- ANVISA của Brazil thực thi việc kiểm tra sinh học tại địa phương
Những khác biệt này buộc các nhà sản xuất phải duy trì 2–3 biến thể thiết bị, làm tăng chi phí phát triển từ 500.000 đến 1,2 triệu USD cho mỗi sản phẩm (Báo cáo Tuân thủ Thiết bị Y tế Toàn cầu 2024).
Xu hướng: Tăng cường mức độ nghiêm ngặt trong các yêu cầu giám sát sau khi lưu hành
Các cơ quan quản lý hiện nay yêu cầu dữ liệu hiệu suất thực tế, với các yêu cầu nghiên cứu sau khi lưu hành của FDA tăng 40% kể từ năm 2021. Các nhà sản xuất phải triển khai hệ thống truy xuất nguồn gốc điện tử để báo cáo sự kiện bất lợi trong vòng 15 ngày – nhanh hơn 50% so với khung thời gian năm 2020. Tỷ lệ thất bại trong các cuộc kiểm toán giám sát đã tăng lên 22% trên toàn ngành, nhấn mạnh nhu cầu quản lý rủi ro chủ động (Tạp chí Quản lý Thiết bị Y tế 2023).
Quản lý chất lượng và chứng nhận sản xuất đối với nhà cung cấp cảm biến IBP
Tầm quan trọng của ISO 13485 trong thiết kế và sản xuất cảm biến IBP
Chứng nhận ISO 13485 thực sự quan trọng đối với quản lý chất lượng trong các nhà cung cấp cảm biến IBP. Nó giúp duy trì kiểm soát ở mọi giai đoạn, từ thiết kế đến sản xuất và cả sau khi sản phẩm ra thị trường. Điều làm nên sự khác biệt so với các tiêu chuẩn chất lượng thông thường là cách nó đáp ứng các yêu cầu cụ thể của thiết bị y tế. Hãy nghĩ đến những yếu tố như khả năng truy xuất nguồn gốc thiết kế, đảm bảo khử trùng hiệu quả, và các quyết định dựa trên rủi ro thực tế thay vì phỏng đoán. Dữ liệu năm 2023 về các thiết bị y tế bị thu hồi cũng cho thấy một điều thú vị: các công ty có chứng nhận ISO 13485 gặp phải khoảng một nửa số vấn đề về tuân thủ tiêu chuẩn so với những công ty không có chứng nhận này. Khi nói riêng về cảm biến IBP, việc tuân thủ các hướng dẫn này đồng nghĩa với việc các cảm biến áp lực và đường dẫn dịch luôn nằm trong giới hạn an toàn nghiêm ngặt và hoạt động ổn định trong suốt vòng đời sử dụng tại các bệnh viện và phòng khám.
Tuân thủ Thực hành Sản xuất Tốt (GMP) trong Sản xuất theo Hợp đồng
Việc tuân thủ các hướng dẫn Thực hành Sản xuất Tốt (GMP) giúp các nhà sản xuất theo hợp đồng duy trì tính nhất quán khi sản xuất các đầu dò IBP đáp ứng cả tiêu chuẩn FDA và EU MDR. Điều gì làm cho GMP trở nên quan trọng đến vậy? Về cơ bản, GMP bao gồm ba lĩnh vực chính: lưu giữ hồ sơ đúng quy định, đảm bảo thiết bị được hiệu chuẩn thường xuyên và đào tạo nhân viên một cách bài bản. Những yếu tố này ảnh hưởng trực tiếp đến độ chính xác của các phép đo trên các thiết bị theo dõi huyết áp xâm lấn. Theo số liệu kiểm toán gần đây, các cơ sở tuân thủ đầy đủ GMP đạt tỷ lệ thành công khoảng 98% trong lần thử nghiệm vô trùng đầu tiên. Con số này cao hơn nhiều so với mức khoảng 72% tại những nơi chỉ tuân thủ một phần các quy định này. Khi các công ty thuê ngoài một phần quy trình sản xuất, ví dụ như chế tạo màng ngăn của đầu dò hoặc lắp ráp chúng, thì việc tuân thủ GMP lại càng trở nên thiết yếu hơn. Những sai sót nhỏ trong quy trình phòng sạch tại các cơ sở bên thứ ba có thể ảnh hưởng nghiêm trọng đến chất lượng sản phẩm cuối cùng.
Câu hỏi thường gặp (FAQ)
Đầu dò IBP thực sự hoạt động như thế nào?
Các bộ chuyển đổi IBP chuyển đổi các chỉ số áp suất từ động mạch thành tín hiệu điện, cho phép theo dõi huyết áp trong thời gian thực trong các thủ tục y tế quan trọng.
Tại sao chứng nhận CE lại quan trọng đối với các bộ chuyển đổi IBP?
Chứng nhận CE đảm bảo rằng các bộ chuyển đổi IBP đáp ứng các tiêu chuẩn an toàn nghiêm ngặt dành cho thiết bị y tế được bán tại châu Âu, từ đó bảo vệ an toàn cho bệnh nhân.
Tiêu chuẩn ISO 10993 hỗ trợ sản xuất bộ chuyển đổi IBP như thế nào?
Các tiêu chuẩn ISO 10993 giúp nhà sản xuất đánh giá tính tương thích sinh học của vật liệu, giảm thiểu các phản ứng sinh học bất lợi khi tiếp xúc với bệnh nhân.
Tổ chức được chỉ định (Notified Bodies) đóng vai trò gì trong việc tuân thủ CE?
Các Tổ chức được chỉ định hoạt động như kiểm toán viên độc lập, xác nhận rằng các bộ chuyển đổi IBP đáp ứng các yêu cầu quy định trước khi đưa ra thị trường.
Tại sao ISO 13485 lại quan trọng đối với quản lý chất lượng trong sản xuất bộ chuyển đổi IBP?
ISO 13485 đảm bảo rằng các nhà cung cấp thiết bị y tế duy trì các tiêu chuẩn cao trong thiết kế, sản xuất và các hoạt động sau khi đưa sản phẩm ra thị trường nhằm đảm bảo chất lượng và độ an toàn của sản phẩm.
Mục Lục
- Hiểu về Cảm biến IBP và Ý nghĩa Lâm sàng của Chúng
- Chứng nhận CE và Tuân thủ EU MDR đối với Cảm biến IBP
-
Tiêu chuẩn Sinh tương thích ISO 10993 và Tiêu chuẩn Quản lý Rủi ro ISO 14971
- Tại sao tiêu chuẩn ISO 10993 lại quan trọng đối với các thành phần tiếp xúc bệnh nhân trong hệ thống IBP
- Đánh giá nguy cơ độc tế bào, dị ứng và kích ứng
- Nghiên cứu điển hình: Những thất bại trong lựa chọn vật liệu dẫn đến vi phạm quy định
- Tích hợp Kiểm tra Tương thích sinh học với Quản lý rủi ro theo ISO 14971
- Phê duyệt FDA và Các yêu cầu quy định toàn cầu đối với cảm biến IBP
- Quản lý chất lượng và chứng nhận sản xuất đối với nhà cung cấp cảm biến IBP
-
Câu hỏi thường gặp (FAQ)
- Đầu dò IBP thực sự hoạt động như thế nào?
- Tại sao chứng nhận CE lại quan trọng đối với các bộ chuyển đổi IBP?
- Tiêu chuẩn ISO 10993 hỗ trợ sản xuất bộ chuyển đổi IBP như thế nào?
- Tổ chức được chỉ định (Notified Bodies) đóng vai trò gì trong việc tuân thủ CE?
- Tại sao ISO 13485 lại quan trọng đối với quản lý chất lượng trong sản xuất bộ chuyển đổi IBP?