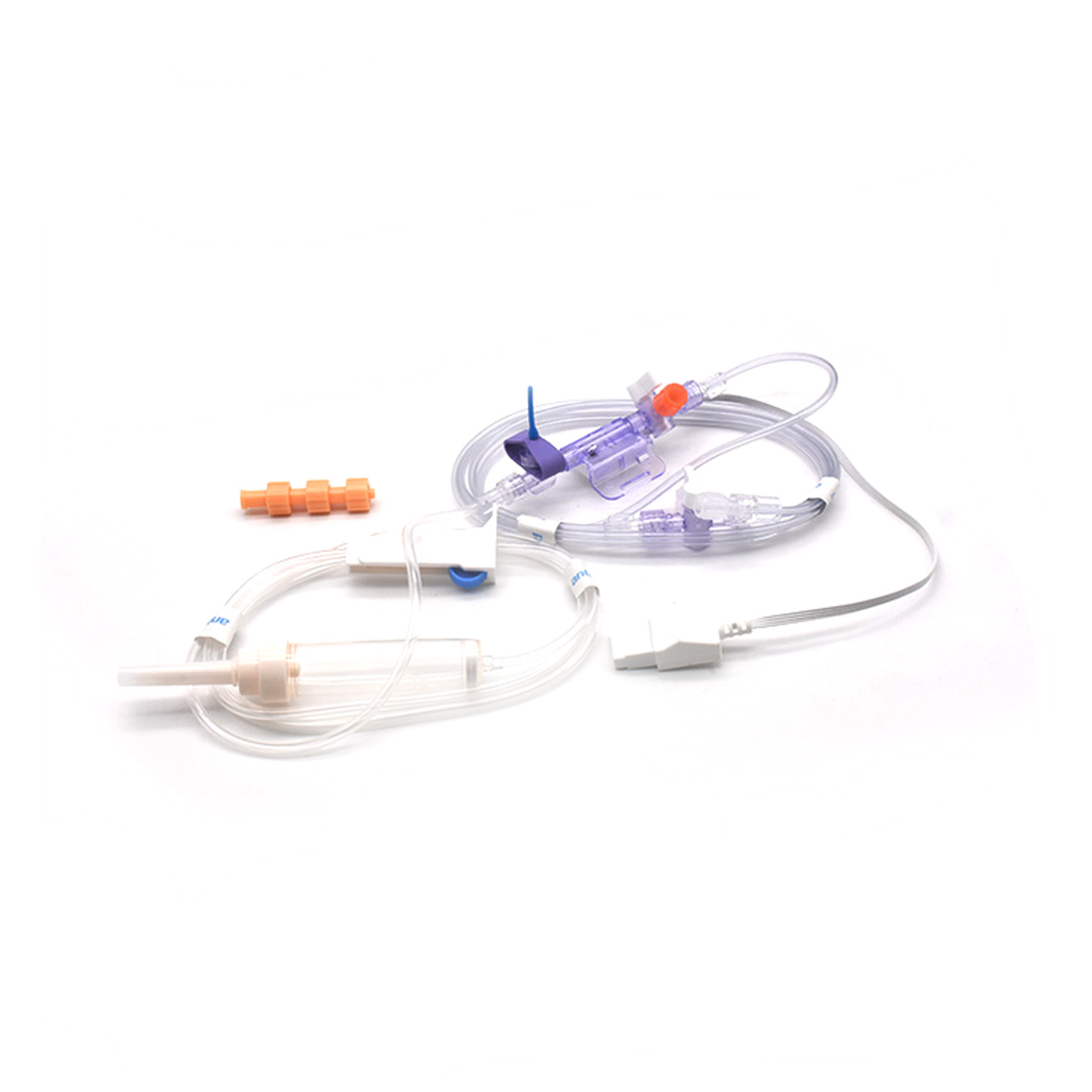
Розуміння трансдюсерів ІАТ та їх клінічне значення
Що таке трансдюктор IBP і як він працює?
Трансдюсери інвазивного вимірювання артеріального тиску (IBP) — це медичні пристрої, які отримують фактичні показники тиску з артерій, підключених через катетери, і перетворюють їх на електричні сигнали, щоб лікарі могли спостерігати за процесами в режимі реального часу. Ці пристрої зазвичай мають сенсори, які зберігаються стерильними, щоб фіксувати навіть найменші зміни рівня тиску, після чого передають усю цю інформацію на монітори через системи, заповнені рідиною. Новіші моделі забезпечують точність близько ±2 мм рт. ст., що має велике значення під час виявлення небезпечних падінь або стрибків артеріального тиску під час операцій. Дослідження, опубліковане минулого року в журналі Journal of Critical Care Medicine, показало цікавий результат: епізоди низького артеріального тиску у пацієнтів, яких спостерігали за допомогою цих точних трансдюсерів, виявлялися на 42% швидше, ніж за допомогою звичайних неінвазивних методів. Таке раннє попередження має велике значення в операційних, де лічаться секунди.
Клінічне значення точного інвазивного моніторингу артеріального тиску
Для пацієнтів, яким потрібні вазопресори або допомога у стабілізації кровообігу, інвазивне вимірювання артеріального тиску (IBP) досі вважається найкращим підходом. Осцилометричні методи не забезпечують достатньої точності, коли потрібні показання щодо кожного серцевого скорочення, особливо важливі під час септичного шоку, після серйозних травм або під час операцій на серці. Лікарі покладаються на швидкість реакції цих моніторів, яка зазвичай становить менше ніж одна секунда, щоб приймати рішення щодо, наприклад, введення рідини. Клінічний досвід показує, що навіть затримка у п’ять хвилин із корекцією проблем з артеріальним тиском може призвести до зростання рівня смертності приблизно на 15%. Нещодавнє дослідження практики роботи в реанімаційних відділеннях показало цікавий результат: лікарні, які впровадили стандартні комплекти датчиків IBP, зафіксували приблизно на 31% менше помилок під час коригування медикаментозного лікування в інтенсивних відділеннях.
Сертифікація СЕ та відповідність вимогам ЄС MDR для датчиків IBP
Що означає сертифікація СЕ для безпеки та ефективності медичних пристроїв
Отримання сертифікації СЕ відповідно до Регламенту ЄС щодо медичних виробів (MDR) означає, що трансдюсер IBP проходить суворі перевірки безпеки, необхідні для продажу в Європі. Позначення СЕ свідчить про те, що продукт відповідає важливим стандартам, таким як IEC 60601 для медичного електронного обладнання, а також виконує правила управління ризиками згідно з ISO 14971. Для пристроїв, що відповідають вимогам MDR, існує весь процес тестування сумісності матеріалів, які контактують з пацієнтами, щоб забезпечити їх безпечне спільне використання та запобігти пошкодженню клітин або подразненням. Більшість пристроїв для моніторингу артеріального тиску належать до категорії IIa або вище, що означає: виробники не можуть самостійно декларувати відповідність, а мають залучити незалежних експертів для підтвердження правильності роботи всіх функцій перед виходом на ринок.
Кроки для отримання позначення СЕ за регламентом ЄС MDR для трансдюсерів IBP
- Класифікувати пристрій : Визначити клас ризику (зазвичай клас IIa для трансдюсерів IBP) згідно з додатком VIII до MDR
- Впровадити систему якості (QMS) : Створіть систему управління якістю, що відповідає стандарту ISO 13485.
- Підготуйте технічну документацію : Збирайте специфікації конструкції, оцінки ризиків та звіти клінічної евалюації.
- Залучіть повідомлений орган : Для пристроїв класу IIa+ пройдіть аудит технічної документації та виробничих процесів.
- Нанесіть знак CE : Після оцінки відповідності зареєструйте пристрій у ЄВДАМЕД. Згідно з даними SimplerQMS, 89% затримок виникають через неповну клінічну евалюацію або застарілу документацію СУЯ.
Роль повідомлених органів у підтвердженні відповідності вимогам CE
Оповіщені органи, або НВ у скороченні, виступають незалежними аудиторами щодо медичних виробів, які класифікуються як високоризикові. Їхнє завдання полягає у перевірці всієї технічної документації та безпосередньому відвідуванні виробничих майданчиків для інспектування. Зокрема щодо трансдюсерів IBP, ці органи перевіряють, чи відповідає все вимогам, викладеним у Додатку II до MDR. Це включає забезпечення належної валідації програмного забезпечення для цифрових версій та дотримання строгих стандартів стерильності для одноразових виробів. Згідно з недавнім опитуванням 2023 року, більшість проблем, виявлених НВ, походять із двох основних сфер: відсутність у компаній надійних стратегій нагляду за продуктами після виходу на ринок або недостатність даних щодо біосумісності. Ці результати показують, на чому саме виробникам слід зосередити увагу, щоб успішно пройти аудит.
Самосертифікація проти оцінювання третіми сторонами для виробів класу IIa
Хоча клас I нестерильних пристроїв може мати самосертифікацію, трансдюсери IBP класу IIa потребують обов’язкового контролю повноваженим органом. Самосертифікація застосовується лише до аксесуарів низького ризику, таких як багаторазові кабелі; компоненти вимірювання тиску потребують повного перегляду третьою стороною. Останні оновлення MDR передбачають щорічні аудити повноважним органом для виробників класу IIa, що зменшило некомп'ютерні виходи на ринок на 34% з 2021 року.
Стандарти біосумісності ISO 10993 та управління ризиками ISO 14971
Чому важливий стандарт ISO 10993 для компонентів, що контактують із пацієнтом, у системах IBP
Для трансдюсерів ІАТ, які фактично контактують із пацієнтами, дотримання стандартів ISO 10993 є майже обов’язковим, якщо ми хочемо уникнути небезпечних біологічних реакцій. Основна мета цього стандарту — визначити, що відбувається, коли людина тривалий час контактує з різноманітними матеріалами, такими як пластикові деталі, клей, що використовується при складанні, та різні сенсорні елементи. Згідно з дослідженням, опублікованим минулого року, приблизно один із п’яти випадків подразнення шкіри, зареєстрованих у лікарнях, був спричинений медичними пристроями, виготовленими з матеріалів, які не відповідають вимогам ISO 10993-10. Компанії, які інтегрують ці вимоги у свій виробничий процес шляхом розробки належного Плану біологічної оцінки, зазвичай стикаються з меншою кількістю проблем, оскільки вони тестують, як різні матеріали реагують при контакті з реальними біологічними рідинами та тканинами протягом часу.
Оцінка ризиків цитотоксичності, сенсибилизації та подразнення
ISO 10993 вимагає проведення трьох основних тестів для матеріалів трансдюсерів ІАТ:
- Цитотоксичність (ISO 10993-5): Визначає життєздатність клітин після впливу матеріалу (допускається загибель ≥70% клітин)
- Сенсибилизація (ISO 10993-10): Оцінює потенційну алергічну реакцію за допомогою тестів з максимальною сенсибилизацією на морських свинках
- Подразнення (ISO 10993-10): Оцінює ризики локального запалення шляхом епідермальних плевкових тестів
Пристрої, що не проходять будь-який тест, мають на 89% вищий рівень вилучення FDA у порівнянні з повністю сумісними аналогами (Звітність про медичні вироби, 2022).
Дослідження випадку: Помилки у виборі матеріалів, що призвели до невідповідності
Аудит ЄС MDR 2021 року показав, що 32% відхилених трансдюсерів IBP використовували силіконові трубки без сертифікації ISO 10993-33 для тривалого контакту з кров'ю. Вилучення виробника на суму 2,7 млн доларів стало наслідком:
- Припущення, що матеріали «медичного класу» автоматично відповідають стандартам біосумісності
- Пропуск прискорених тестів старіння щодо ризиків виділення хімічних речовин
- Недостатня документація впливу стерилізації на безпеку матеріалів
Інтеграція випробувань біосумісності з управлінням ризиками відповідно до ISO 14971
Поєднання даних ISO 10993 з рамками оцінки ризиків ISO 14971 дозволяє виробникам:
- Кількісно оцінювати біологічні ризики за допомогою матриць тяжкості/ймовірності небезпек
- Застосовувати заходи контролю, такі як заміна матеріалів або нанесення біосумісних покриттів
- Відстежувати небажані події після виходу на ринок через процеси, узгоджені з ISO 13485
Такий подвійний підхід скорочує кількість змін конструкції на пізніх етапах на 41% порівняно з окремими випробуваннями біосумісності (MedTech Quality Benchmark, 2023).
Схвалення FDA та глобальні регуляторні вимоги для перетворювачів IBP
Процес схвалення FDA 510(k) та суттєва еквівалентність
Для виробників медичних приладів отримання дозволу через програму FDA 510(k) означає демонстрацію того, що їхній продукт суттєво еквівалентний уже наявному на ринку. У 2023 році близько трьох чвертей усіх пристроїв було схвалено саме цим шляхом, згідно з даними галузі. Виконання цих вимог потребує серйозного обсягу випробувань та суворого дотримання правил якісних систем, встановлених у 21 CFR Part 820. Компанії, які мають сертифікацію ISO 13485, як правило, проходять процес розгляду заявок у FDA приблизно на 30 відсотків швидше. Це, здається, пояснюється кращою організацією документації та практик документування, які відповідають очікуванням регуляторів під час перевірки.
Виклики гармонізації: відмінності між ЄС MDR, FDA та іншими ринками
Розбіжності в регіональних вимогах створюють складність у дотриманні норм:
- ЄС MDR передбачає клінічну оцінку для отримання знаку СЕ
- FDA акцентує увагу на основі прецедентної еквівалентності
- Японське PMDA вимагає відповідності ISO 80369-6 для нейровісцеральних з'єднань
- Національна санітарна служба Бразилії (ANVISA) вимагає проведення локального тестування на біосумісність
Ці розбіжності змушують виробників підтримувати 2–3 варіанти пристроїв, що збільшує витрати на розробку на 500 тис. – 1,2 млн доларів США на продукт (Звіт Global MedTech Compliance Report, 2024).
Тенденція: посилення вимог до нагляду за продуктами після виходу на ринок
Регулятори тепер вимагають дані про реальну ефективність виробів; кількість досліджень, які FDA вимагає проводити після виходу на ринок, зросла на 40% з 2021 року. Виробники мають впроваджувати електронні системи просування для повідомлення про небажані події протягом 15 днів — на 50% швидше, ніж у 2020 році. Рівень невдалих аудитів нагляду зріс до 22% у галузі загалом, що підкреслює необхідність проактивного управління ризиками (Medical Device Regulatory Journal, 2023).
Системи управління якістю та виробничі сертифікації для постачальників трансдюсерів IBP
Важливість стандарту ISO 13485 у проектуванні та виробництві трансдюсерів IBP
Сертифікація за ISO 13485 має велике значення для управління якістю серед постачальників трансдюсерів IBP. Вона допомагає підтримувати контроль на всіх етапах — від проектування до виробництва та подальшої експлуатації після виходу продуктів на ринок. Те, що відрізняє цей стандарт від звичайних вимог до якості, — це врахування специфічних потреб медичних приладів. Мається на увазі можливість відстеження проектів від початку їхньої розробки, забезпечення належного стерилізаційного процесу та прийняття рішень на основі реальних ризиків, а не припущень. Аналіз даних за 2023 рік щодо вилучених з ринку медичних приладів також демонструє цікавий результат: компанії, які мають сертифікацію ISO 13485, мали приблизно вдвічі менше порушень вимог у порівнянні з тими, хто її не має. Коли мова йде про трансдюсери IBP, дотримання цих рекомендацій означає, що датчики тиску та шляхи руху рідини залишаються в межах суворих норм безпеки та надійно працюють протягом усього життєвого циклу в лікарнях та клініках.
Відповідність правилам доброго виробничого контролю (GMP) у контрактному виробництві
Дотримання рекомендацій щодо доброго виробничого контролю (GMP) допомагає договірним виробникам забезпечувати стабільність при виготовленні трансдюсерів ІАТ, які відповідають стандартам FDA та ЄС MDR. Чому GMP так важлива? По-перше, вона охоплює три основні аспекти: належне ведення документації, забезпечення калібрування обладнання та правильну підготовку персоналу. Ці фактори безпосередньо впливають на точність вимірювань інвазивних моніторів артеріального тиску. Згідно з останніми даними перевірок, підприємства, які повністю дотримуються GMP, досягають близько 98% успішних результатів під час першої спроби тестів на стерильність. Це значно краще, ніж приблизно 72%, зафіксовані у закладах, які лише частково дотримуються цих правил. Коли компанії передають на аутсорсинг частину виробничого процесу, наприклад, виготовлення діафрагм трансдюсера чи їх складання, дотримання GMP стає ще важливішим. Невеликі помилки в чистих кімнатах на таких сторонніх підприємствах можуть серйозно погіршити якість кінцевого продукту.
Часто задані питання (FAQ)
Що саме робить трансдюсер ІАТ?
Трансдюсери IBP перетворюють показання тиску з артерій на електричні сигнали, що дозволяє в режимі реального часу контролювати артеріальний тиск під час критичних медичних процедур.
Чому важлива сертифікація СЕ для трансдюсерів IBP?
Сертифікація СЕ гарантує, що трансдюсери IBP відповідають суворим стандартам безпеки, необхідним для медичних приладів, що продаються в Європі, забезпечуючи безпеку пацієнтів.
Як стандарти ISO 10993 допомагають у виробництві трансдюсерів IBP?
Стандарти ISO 10993 дають змогу виробникам оцінювати матеріали на біосумісність, мінімізуючи небажані біологічні реакції при контакті з пацієнтом.
Яку роль відіграють повідомлені органи у відповідності вимогам СЕ?
Повідомлені органи виступають як незалежні аудитори, які підтверджують, що трансдюсери IBP відповідають регуляторним вимогам перед виходом на ринок.
Чому важливий стандарт ISO 13485 для системи управління якістю при виробництві трансдюсерів IBP?
ISO 13485 забезпечує, щоб постачальники медичних приладів дотримувалися високих стандартів на всіх етапах — від проектування до виробництва та післяреалізаційної діяльності — задля забезпечення якості та безпеки продуктів.
Зміст
- Розуміння трансдюсерів ІАТ та їх клінічне значення
- Сертифікація СЕ та відповідність вимогам ЄС MDR для датчиків IBP
-
Стандарти біосумісності ISO 10993 та управління ризиками ISO 14971
- Чому важливий стандарт ISO 10993 для компонентів, що контактують із пацієнтом, у системах IBP
- Оцінка ризиків цитотоксичності, сенсибилизації та подразнення
- Дослідження випадку: Помилки у виборі матеріалів, що призвели до невідповідності
- Інтеграція випробувань біосумісності з управлінням ризиками відповідно до ISO 14971
- Схвалення FDA та глобальні регуляторні вимоги для перетворювачів IBP
- Системи управління якістю та виробничі сертифікації для постачальників трансдюсерів IBP
-
Часто задані питання (FAQ)
- Що саме робить трансдюсер ІАТ?
- Чому важлива сертифікація СЕ для трансдюсерів IBP?
- Як стандарти ISO 10993 допомагають у виробництві трансдюсерів IBP?
- Яку роль відіграють повідомлені органи у відповідності вимогам СЕ?
- Чому важливий стандарт ISO 13485 для системи управління якістю при виробництві трансдюсерів IBP?