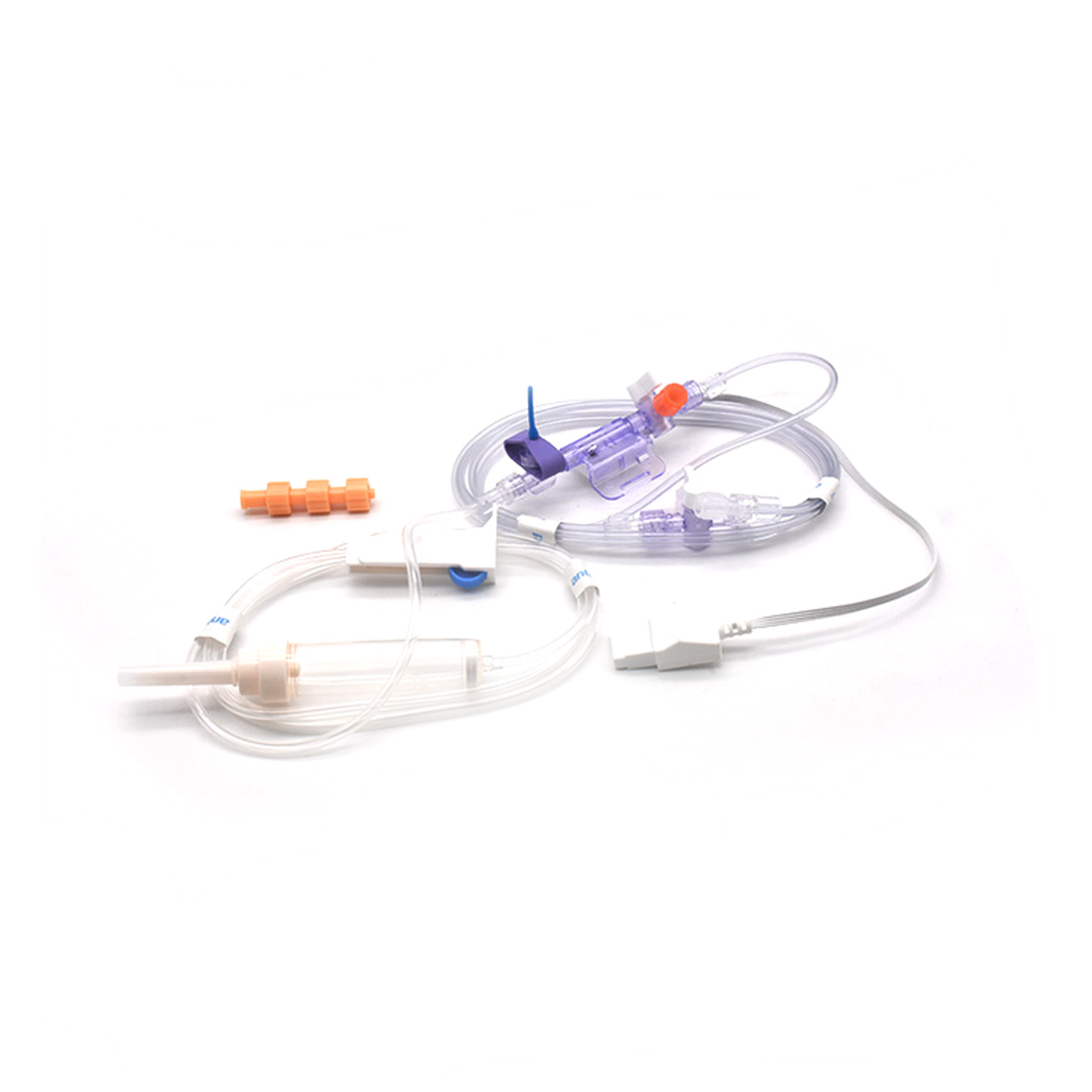
Понимание датчиков ИАД и их клиническое значение
Что такое IBP-трансдуктор и как он работает?
Датчики инвазивного артериального давления (IBP) — это медицинские устройства, которые снимают фактические показания давления из артерий, подключённых через катетеры, и преобразуют их в электрические сигналы, чтобы врачи могли отслеживать происходящее в режиме реального времени. Эти устройства обычно оснащены стерильными сенсорами, способными улавливать даже самые незначительные изменения уровня давления, после чего передают всю эту информацию на мониторы посредством заполненных жидкостью систем. Современные модели обеспечивают точность около ±2 мм рт.ст., что особенно важно при выявлении опасных падений или скачков артериального давления во время операций. Исследование, опубликованное в прошлом году в журнале «Journal of Critical Care Medicine», показало интересный результат: эпизоды низкого артериального давления у пациентов, находящихся под наблюдением с помощью этих точных датчиков, выявлялись на 42 % быстрее по сравнению с обычными неинвазивными методами. Такое раннее предупреждение имеет большое значение в операционных, где каждая секунда на счету.
Клиническая значимость точного инвазивного мониторинга артериального давления
Для пациентов, которым требуются вазопрессоры или помощь в стабилизации кровообращения, инвазивный мониторинг артериального давления (IBP) по-прежнему считается наилучшим методом. Осциллометрические методы не подходят, когда нам нужны показания с точностью до каждого сердечного сокращения, особенно важные при септическом шоке, после серьезных травм или во время операций на сердце. Врачи полагаются на быструю реакцию этих мониторов — как правило, менее одной секунды — чтобы принимать решения, например, о введении жидкости. Клинический опыт показывает, что даже задержка в пять минут с коррекцией проблем с артериальным давлением может привести к увеличению смертности примерно на 15%. Недавний анализ практик в отделениях интенсивной терапии выявил интересную закономерность: больницы, внедрившие стандартные настройки датчиков IBP, зафиксировали примерно на 31% меньше ошибок при корректировке медикаментозной терапии в своих отделениях интенсивной помощи.
Сертификация СЕ и соответствие требованиям Европейского регламента по медицинским изделиям (EU MDR) для датчиков IBP
Что означает сертификация СЕ для безопасности и эффективности медицинских изделий
Получение сертификата СЕ в соответствии с Регламентом ЕС по медицинским изделиям (MDR) означает, что датчик ИАД проходит строгие проверки безопасности, необходимые для продажи в Европе. Знак СЕ подтверждает, что продукт соответствует важным стандартам, таким как IEC 60601 для медицинской электронной аппаратуры, а также соблюдает правила управления рисками по ISO 14971. Для устройств, соответствующих требованиям MDR, предусмотрен процесс тестирования биосовместимости материалов, контактирующих с пациентом, чтобы убедиться, что они не вызывают повреждения клеток или раздражения. Большинство устройств для мониторинга артериального давления относятся к классу IIa или выше, что означает, что производители не могут самостоятельно декларировать соответствие, а должны привлечь независимых экспертов для проверки работоспособности перед выходом на рынок.
Этапы получения знака СЕ по регламенту ЕС MDR для датчиков ИАД
- Классифицируйте устройство : Определите класс риска (обычно класс IIa для датчиков ИАД) в соответствии с Приложением VIII MDR.
- Внедрите систему менеджмента качества (QMS) : Создание системы управления качеством, сертифицированной по стандарту ISO 13485.
- Подготовка технической документации : Составление спецификаций на конструкцию, оценок рисков и отчётов по клинической оценке.
- Привлечение уполномоченного органа : Для изделий класса IIa и выше — прохождение аудита технической документации и производственных процессов.
- Нанесение знака СЕ : После оценки соответствия регистрация изделия в EUDAMED. По данным SimplerQMS, 89 % задержек вызваны неполными клиническими оценками или устаревшей документацией системы управления качеством.
Роль уполномоченных органов при подтверждении соответствия требованиям СЕ
Уведомленные органы, или сокращенно NB, выступают в качестве независимых аудиторов при оценке медицинских изделий, отнесенных к категории повышенного риска. Их задача заключается в тщательной проверке всей технической документации, а также в проведении инспекций на производственных площадках. Что касается датчиков IBP, эти органы проверяют соответствие всех требований, изложенных в Приложении II MDR. Это включает в себя обеспечение надлежащей валидации программного обеспечения для цифровых версий и соблюдение строгих стандартов стерильности одноразовых изделий. Согласно недавнему опросу 2023 года, большинство проблем, выявленных уведомленными органами, связано с двумя основными областями: отсутствием у компаний четких стратегий постмаркетингового наблюдения или недостаточностью данных по биосовместимости. Эти результаты подчеркивают направления, на которых производителям необходимо сосредоточить внимание для успешного прохождения аудита.
Самостоятельная сертификация против оценки третьей стороной для изделий класса IIa
Хотя изделия класса I без стерильной обработки могут подтверждать соответствие самостоятельно, трансдьюсеры для измерения артериального давления класса IIa требуют обязательного контроля уполномоченным органом. Самостоятельное подтверждение применимо только к низкорисковым аксессуарам, таким как многоразовые кабели; компоненты, чувствительные к давлению, требуют полной проверки третьей стороной. Согласно последним обновлениям MDR, производители изделий класса IIa обязаны проходить ежегодные аудиты уполномоченными органами, что снизило количество недобросовестных выходов на рынок на 34 % с 2021 года.
Стандарты биосовместимости ISO 10993 и управления рисками ISO 14971
Почему стандарт ISO 10993 важен для компонентов, контактирующих с пациентом, в системах измерения артериального давления
Для трансдьюсеров ИАД, которые фактически соприкасаются с пациентами, соблюдение стандартов ISO 10993 практически обязательно, если мы хотим избежать нежелательных биологических реакций. Основная цель этого стандарта — определить, что происходит, когда люди в течение длительного времени подвергаются воздействию различных материалов, таких как пластиковые детали, клей, используемый при сборке, и различные элементы датчиков. Согласно исследованию, опубликованному в прошлом году, около одного из пяти случаев раздражения кожи, зарегистрированных в больницах, были вызваны медицинскими изделиями, изготовленными из материалов, не соответствующих требованиям ISO 10993-10. Компании, которые внедряют эти требования в производственный процесс посредством надлежащих Планов биологической оценки, сталкиваются с меньшим количеством проблем, поскольку они тестируют, как различные материалы реагируют при контакте с реальными биологическими жидкостями и тканями со временем.
Оценка рисков цитотоксичности, сенсибилизации и раздражения
ISO 10993 требует проведения трех основных испытаний для материалов трансдьюсеров ИАД:
- Цитотоксичность (ISO 10993-5): Оценивает жизнеспособность клеток после воздействия материала (допускается гибель до 70% клеток)
- Сенсибилизация (ISO 10993-10): Оценивает потенциальную аллергическую реакцию с использованием тестов максимизации на морских свинках
- Ирритация (ISO 10993-10): Оценивает риски локального воспаления с помощью аппликационных тестов на эпидермисе
Устройства, не прошедшие хотя бы один тест, имеют на 89% более высокий риск отзыва FDA по сравнению с полностью соответствующими требованиям аналогами (Medical Device Reporting, 2022).
Пример из практики: ошибки в выборе материалов, приведшие к несоответствию требованиям
Аудит ЕС по MDR в 2021 году показал, что 32% отвергнутых датчиков IBP использовали силиконовые трубки, не имеющие сертификата ISO 10993-33 для длительного контакта с кровью. Причиной отзыва производителя на сумму 2,7 млн долларов США стали:
- Предположение, что материалы «медицинского класса» автоматически соответствуют стандартам биосовместимости
- Пропуск ускоренных испытаний на предмет рисков выщелачивания химических веществ
- Недостаточная документация по влиянию стерилизации на безопасность материалов
Интеграция испытаний на биосовместимость с управлением рисками в соответствии с ISO 14971
Сочетание данных ISO 10993 с системой управления рисками по ISO 14971 позволяет производителям:
- Количественно оценивать биологические риски с использованием матриц тяжести/вероятности опасностей
- Внедрять меры контроля, такие как замена материалов или нанесение биосовместимых покрытий
- Отслеживать нежелательные явления после выхода на рынок с помощью процессов, соответствующих ISO 13485
Такой двойной подход снижает количество изменений конструкции на поздних этапах на 41 % по сравнению с изолированными испытаниями на биосовместимость (MedTech Quality Benchmark, 2023).
Сертификация FDA и глобальные нормативные требования к датчикам IBP
Процесс получения разрешения FDA 510(k) и подтверждение существенного сходства
Для производителей медицинских изделий получение разрешения через программу FDA 510(k) означает демонстрацию того, что их продукт в значительной степени эквивалентен уже существующему на рынке образцу. В 2023 году около трех четвертей всех устройств были одобрены именно этим способом, согласно отраслевым данным. Соответствие этим требованиям требует серьезных испытаний и строгого соблюдения правил системы качества, изложенных в 21 CFR Часть 820. У компаний, имеющих сертификацию ISO 13485, заявки, как правило, рассматриваются FDA примерно на 30 процентов быстрее. Это, по-видимому, связано с более организованной подачей документации и практиками оформления документов, соответствующими тому, что регуляторы ожидают увидеть при проверке.
Проблемы гармонизации: различия между ЕС MDR, FDA и другими рынками
Различающиеся региональные требования создают сложности в обеспечении соответствия:
- ЕС MDR требует проведения клинической оценки для получения знака СЕ
- FDA делает акцент на эквивалентности по отношению к предшествующему образцу (predicate-based equivalence)
- Японское PMDA требует соответствия ISO 80369-6 для нейроосевых соединений
- Бразильское агентство ANVISA требует проведения локальных испытаний на биосовместимость
Эти расхождения вынуждают производителей поддерживать 2–3 варианта устройств, что увеличивает расходы на разработку на 500 тыс. – 1,2 млн долларов США на продукт (Глобальный отчет по соответствию в сфере медицинских технологий, 2024).
Тенденция: ужесточение требований к надзору за продуктами на рынке
Регулирующие органы теперь требуют данные о реальной эффективности продукции; требования FDA к пострегистрационным исследованиям увеличились на 40% с 2021 года. Производители обязаны внедрять электронные системы прослеживаемости для сообщения о нежелательных явлениях в течение 15 дней — на 50% быстрее, чем сроки 2020 года. Уровень несоответствий по результатам проверок надзора вырос до 22% в масштабах отрасли, что подчеркивает необходимость проактивного управления рисками (Журнал регулирования медицинских изделий, 2023).
Системы управления качеством и производственные сертификаты для поставщиков трансдьюсеров ИАД
Важность стандарта ISO 13485 при разработке и производстве трансдьюсеров ИАД
Сертификация по ISO 13485 имеет большое значение для управления качеством среди поставщиков датчиков ИАД. Она помогает поддерживать контроль на всех этапах — от проектирования до производства и последующего поведения продукции после выхода на рынок. То, что отличает этот стандарт от обычных требований к качеству, — это учёт специфических потребностей медицинских изделий. Речь идёт о таких аспектах, как возможность прослеживания проектных решений до их источника, обеспечение правильной стерилизации и принятие решений на основе фактических рисков, а не предположений. Анализ данных за 2023 год по отзыву медицинских изделий также показал интересную тенденцию: компании, имеющие сертификат ISO 13485, сталкивались с нарушениями стандартов примерно вдвое реже, чем те, у кого его нет. Что касается датчиков ИАД, соблюдение этих руководящих принципов означает, что датчики давления и пути движения жидкости остаются в строгих пределах безопасности и надёжно работают на протяжении всего жизненного цикла в больницах и клиниках.
Соблюдение правил надлежащей производственной практики (GMP) в контрактном производстве
Соблюдение рекомендаций по надлежащей производственной практике (GMP) помогает подрядным производителям обеспечивать стабильность при изготовлении трансдьюсеров ИАД, соответствующих стандартам FDA и ЕС MDR. Почему GMP так важна? Дело в том, что она охватывает три основные области: ведение надлежащей документации, обеспечение калибровки оборудования и правильную подготовку персонала. Эти аспекты напрямую влияют на точность измерений инвазивных мониторов артериального давления. Согласно последним данным аудита, предприятия, полностью соблюдающие GMP, достигают примерно 98% успеха при первом прохождении тестов на стерильность. Это намного выше, чем примерно 72%, наблюдаемые на предприятиях, лишь частично следующих этим правилам. Когда компании передают на аутсорсинг части производственного процесса, такие как создание диафрагм трансдьюсера или их сборка, соблюдение GMP становится ещё более важным. Небольшие ошибки в процедурах чистых помещений на этих сторонних площадках могут серьёзно повлиять на качество конечного продукта.
Часто задаваемые вопросы (FAQ)
Что именно делает трансдьюсер ИАД?
Датчики ИАД преобразуют показания давления из артерий в электрические сигналы, обеспечивая мониторинг артериального давления в режиме реального времени во время критически важных медицинских процедур.
Почему сертификация СЕ важна для датчиков ИАД?
Сертификация СЕ гарантирует, что датчики ИАД соответствуют строгим стандартам безопасности, необходимым для медицинских устройств, продаваемых в Европе, и обеспечивает безопасность пациентов.
Как стандарты ISO 10993 помогают в производстве датчиков ИАД?
Стандарты ISO 10993 позволяют производителям оценивать материалы на биосовместимость, минимизируя нежелательные биологические реакции при контакте с пациентом.
Какую роль играют уполномоченные органы в соответствии требованиям СЕ?
Уполномоченные органы выступают в качестве независимых аудиторов, подтверждающих соответствие датчиков ИАД регуляторным требованиям перед выходом на рынок.
Почему стандарт ISO 13485 важен для системы управления качеством при производстве датчиков ИАД?
ISO 13485 гарантирует, что поставщики медицинских устройств поддерживают высокие стандарты на всех этапах — от разработки и производства до послепродажного обслуживания — для обеспечения качества и безопасности продукции.
Содержание
- Понимание датчиков ИАД и их клиническое значение
-
Сертификация СЕ и соответствие требованиям Европейского регламента по медицинским изделиям (EU MDR) для датчиков IBP
- Что означает сертификация СЕ для безопасности и эффективности медицинских изделий
- Этапы получения знака СЕ по регламенту ЕС MDR для датчиков ИАД
- Роль уполномоченных органов при подтверждении соответствия требованиям СЕ
- Самостоятельная сертификация против оценки третьей стороной для изделий класса IIa
-
Стандарты биосовместимости ISO 10993 и управления рисками ISO 14971
- Почему стандарт ISO 10993 важен для компонентов, контактирующих с пациентом, в системах измерения артериального давления
- Оценка рисков цитотоксичности, сенсибилизации и раздражения
- Пример из практики: ошибки в выборе материалов, приведшие к несоответствию требованиям
- Интеграция испытаний на биосовместимость с управлением рисками в соответствии с ISO 14971
- Сертификация FDA и глобальные нормативные требования к датчикам IBP
- Системы управления качеством и производственные сертификаты для поставщиков трансдьюсеров ИАД
-
Часто задаваемые вопросы (FAQ)
- Что именно делает трансдьюсер ИАД?
- Почему сертификация СЕ важна для датчиков ИАД?
- Как стандарты ISO 10993 помогают в производстве датчиков ИАД?
- Какую роль играют уполномоченные органы в соответствии требованиям СЕ?
- Почему стандарт ISO 13485 важен для системы управления качеством при производстве датчиков ИАД?