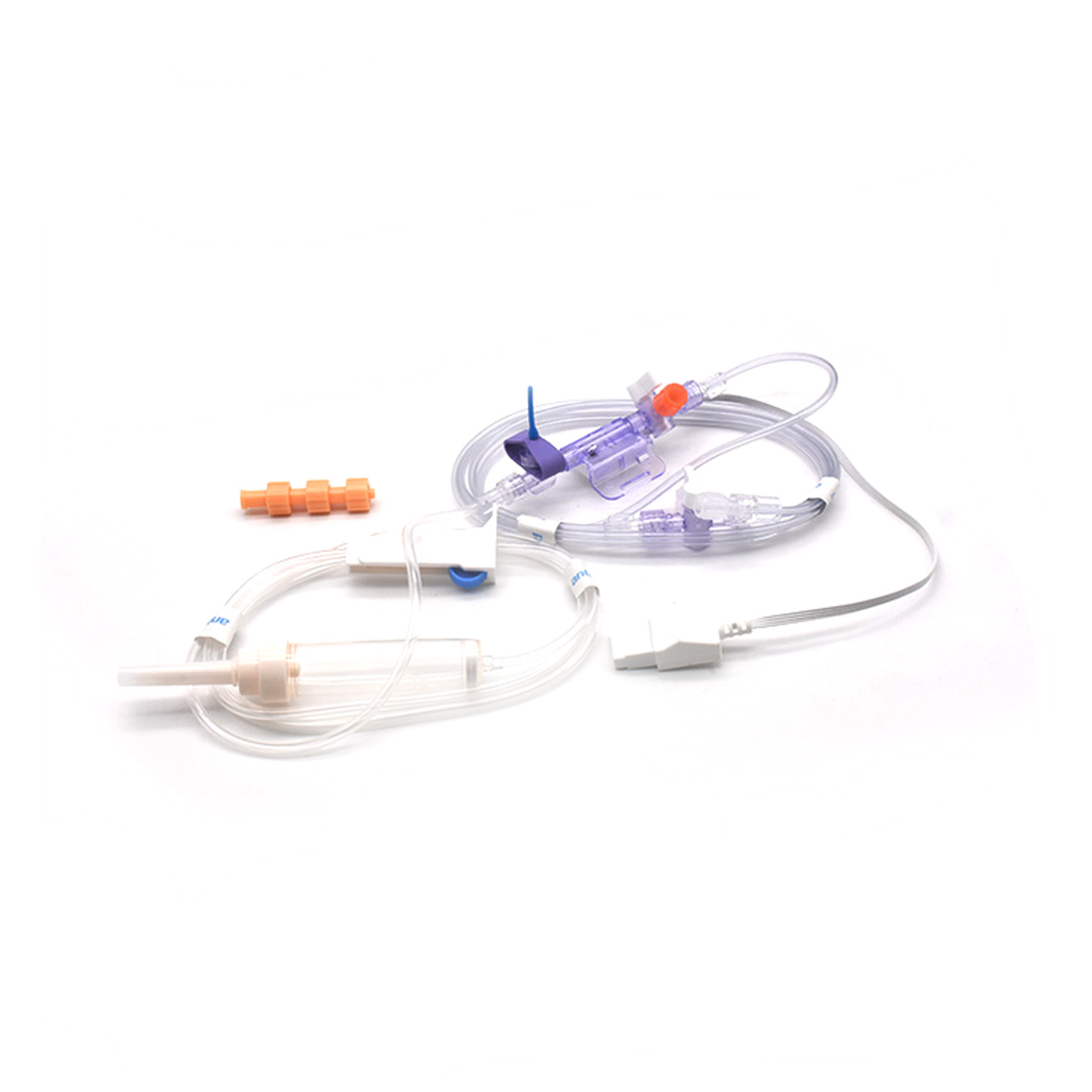
Înțelegerea transductoarelor IBP și importanța lor clinică
Ce este un transductor IBP și cum funcționează?
Transductoarele de presiune arterială invazivă (IBP) sunt acele dispozitive medicale care iau măsurători reale ale presiunii din artere prin intermediul cateterelor și le transformă în semnale electrice, astfel încât medicii să poată urmări în timp real ce se întâmplă. Aceste dispozitive au în general senzori menținuți sterili pentru a detecta chiar și cele mai mici variații ale nivelului de presiune, apoi transmit toate aceste informații la monitoare prin sisteme umplute cu lichid. Modelele mai noi ating o precizie de aproximativ ±2 mmHg, ceea ce este esențial pentru detectarea scăderilor sau creșterilor periculoase ale tensiunii arteriale în timpul operațiilor. O cercetare publicată anul trecut în Journal of Critical Care Medicine a evidențiat un aspect interesant – pacienții monitorizați cu aceste transductoare precise au avut episoadele de hipotensiune depistate cu 42% mai rapid decât prin tehnici non-invazive obișnuite. Un avertizament atât de precoce face o mare diferență în sălile de operație, unde fiecare secundă contează.
Importanța clinică a monitorizării precise a presiunii arteriale invazive
Pentru pacienții care au nevoie de vasopresoare sau ajutor pentru stabilizarea circulației, monitorizarea invazivă a tensiunii arteriale (IBP) este încă considerată cea mai bună abordare. Metodele oscilometrice nu sunt suficiente atunci când avem nevoie de măsurători beat-cu-beat, atât de importante în cazurile de oc septic, după accidente majore sau în timpul operațiilor cardiace. Medicii se bazează pe viteza de răspuns a acestor monitoare, de obicei sub o secundă, pentru a decide aspecte precum administrarea de lichide. Am observat din experiența clinică că chiar și așteptarea a cinci minute întregi pentru corectarea problemelor de tensiune arterială poate duce la o rată a mortalității cu aproximativ 15% mai mare. O analiză recentă a practicilor din secțiile de terapie intensivă a arătat și un alt aspect interesant: spitalele care au implementat setări standardizate de transductori IBP au înregistrat cu aproximativ 31% mai puține erori la ajustarea medicamentelor în secțiile lor de terapie intensivă.
Certificare CE și conformitate cu reglementarea UE MDR pentru transductori IBP
Ce înseamnă certificarea CE pentru siguranța și performanța dispozitivelor medicale
Obținerea certificării CE conform Regulamentului Uniunii Europene privind dispozitivele medicale (MDR) înseamnă, în esență, că un traductor IBP trece de verificări riguroase de siguranță necesare pentru a fi vândut în Europa. Marcarea CE arată că produsul respectă standarde importante precum IEC 60601 pentru echipamentele electronice medicale și că îndeplinește, de asemenea, regulile de management al riscurilor din ISO 14971. Pentru dispozitivele care respectă reglementările MDR, există întregul proces de testare a compatibilității materialelor care vin în contact cu pacienții, asigurându-se că acestea nu vor provoca leziuni celulare sau probleme de iritație. Majoritatea dispozitivelor de monitorizare a tensiunii arteriale se încadrează în categoria Clasa IIa sau superioară, ceea ce înseamnă că producătorii nu pot declara conformitatea doar prin autodeclarare, ci trebuie să implice experți independenți pentru a verifica dacă totul funcționează corespunzător înainte de lansarea pe piață.
Pași pentru obținerea marcării CE în conformitate cu MDR UE pentru traductoarele IBP
- Clasificați dispozitivul : Determinați clasificarea riscului (în mod tipic Clasa IIa pentru traductoarele IBP) conform Anexei VIII MDR.
- Implementați un SMC : Stabiliți un sistem de management al calității certificat ISO 13485.
- Pregătiți documentația tehnică : Compilați specificațiile de proiectare, evaluările de risc și rapoartele de evaluare clinică.
- Implicați un Organism Notificat : Pentru dispozitivele de clasa IIa+, efectuați audituri ale dosarelor tehnice și proceselor de fabricație.
- Aplicați marcajul CE : După evaluarea conformității, înregistrați dispozitivul în EUDAMED. Potrivit SimplerQMS, 89% dintre întârzieri provin din evaluări clinice incomplete sau din documentația QMS neactualizată.
Rolul Organismelor Notificate în validarea conformității CE
Organismele notificate, sau ON, scurt, acționează ca auditori independenți în cazul dispozitivelor medicale clasificate ca având un risc mai mare. Activitatea lor presupune analizarea tuturor documentelor tehnice și prezența efectivă în unitățile de producție pentru inspecții. În ceea ce privește transductoarele IBP, aceste organisme verifică dacă toate aspectele corespund cerințelor prevăzute în Anexa II din MDR. Aceasta include asigurarea că versiunile digitale au o validare adecvată a software-ului și că produsele utilizabile o singură dată respectă standarde stricte de sterilitate. Conform unui sondaj recent din 2023, cele mai multe probleme identificate de ON provin din două domenii principale: lipsa unor strategii solide de supraveghere post-marketing din partea companiilor sau incapacitatea de a furniza date suficiente privind biocompatibilitatea. Aceste constatări subliniază zonele la care trebuie să se concentreze producătorii dacă doresc să treacă cu succes auditurile.
Autocertificare vs. Evaluare de către terți pentru dispozitivele din clasa IIa
Deși dispozitivele de clasa I non-sterile se pot autocertifica, transductoarele IBP de clasa IIa necesită supraveghere obligatorie din partea unui organism notificat. Autocertificarea se aplică doar accesoriilor cu risc scăzut, cum ar fi cablurile reutilizabile; componentele senzor de presiune necesită o verificare completă de către o parte terță. Actualizările recente ale MDR prevăd audituri anuale ale organismului notificat pentru producătorii de clase IIa, reducând intrările neconforme pe piață cu 34% începând din 2021.
Standardele ISO 10993 privind biocompatibilitatea și ISO 14971 privind managementul riscurilor
De ce este importantă norma ISO 10993 pentru componentele care vin în contact cu pacientul în sistemele IBP
Pentru transductoarele IBP care intră în contact direct cu pacienții, urmarea standardelor ISO 10993 este practic obligatorie dacă vrem să evităm acele reacții biologice neplăcute. Scopul acestui standard este de a determina ce se întâmplă atunci când oamenii sunt expuși pe perioade lungi la diverse materiale, cum ar fi piese din plastic, adezivi utilizați în asamblare și diferite componente ale senzorilor. Conform unui studiu publicat anul trecut, aproximativ unul din cinci cazuri de iritații ale pielii raportate în spitale proveneau de la dispozitive medicale fabricate cu materiale care nu respectau cerințele ISO 10993-10. Companiile care integrează aceste aspecte în procesul lor de producție prin planuri adecvate de evaluare biologică întâmpină mai puține probleme, deoarece testează modul în care diferitele materiale reacționează atunci când vin în contact cu fluide și țesuturi corporale reale pe durată.
Evaluarea riscurilor de citotoxicitate, sensibilizare și iritație
ISO 10993 necesită trei teste de bază pentru materialele transductoarelor IBP:
- Citotoxicitate (ISO 10993-5): Măsoară viabilitatea celulelor după expunerea la material (permisă moarte celulară de până la 70%)
- Sensibilizare (ISO 10993-10): Evaluează potențialul de reacție alergică folosind testele maximale pe cobai
- Iritare (ISO 10993-10): Evaluează riscurile de inflamație localizată prin teste cutanate epidermice
Dispozitivele care nu trec oricare dintre testele au un risc cu 89% mai mare de a fi retrase de către FDA comparativ cu alternativele complet conforme (Raportarea Dispozitivelor Medicale, 2022).
Studiu de caz: Eșecuri în selecția materialelor care au condus la neconformitate
Un audit EU MDR din 2021 a relevat că 32% dintre transductoarele IBP respinse utilizau tuburi din silicon care nu aveau certificare ISO 10993-33 pentru contact sanguin prelungit. Retragerea producătorului în valoare de 2,7 milioane USD a rezultat din:
- Presupunerea că materialele "de uz medical" îndeplineau automat standardele de biocompatibilitate
- Săritul testelor de îmbătrânire accelerată pentru riscurile de eliberare chimică
- Documentare inadecvată a impactului sterilizării asupra siguranței materialelor
Integrarea testării biocompatibilității cu managementul riscurilor conform ISO 14971
Combinarea datelor din ISO 10993 cu cadrul de risc al ISO 14971 permite producătorilor să:
- Cuantifice riscurile biologice utilizând matrice de gravitate/probabilitate a pericolelor
- Implementeze controale precum înlocuirea materialelor sau aplicarea unor straturi protectoare biocompatibile
- Monitorizeze evenimentele adverse post-marketing prin procese conforme cu ISO 13485
Această abordare duală reduce modificările de design din fazele avansate cu 41% comparativ cu testarea izolată a biocompatibilității (Referință calitate MedTech, 2023).
Aprobarea FDA și cerințele reglementare globale pentru traductoarele IBP
Procesul de aprobare FDA 510(k) și echivalența substanțială
Pentru producătorii de dispozitive medicale, obținerea autorizării prin programul FDA 510(k) înseamnă demonstrarea faptului că produsul lor este substanțial echivalent cu un produs deja existent pe piață. Aproximativ trei sferturi din toate dispozitivele au fost aprobate în acest fel în 2023, conform datelor din industrie. Îndeplinirea acestor cerințe necesită teste riguroase și respectarea strictă a regulilor 21 CFR Part 820 privind sistemele de calitate. Companiile care dețin certificarea ISO 13485 tind să își vadă cererile procesate cu aproximativ 30 la sută mai rapid de către FDA. Acest lucru pare să se datoreze unor practici mai bine organizate de întocmire a documentației, care corespund așteptărilor reglementare în timpul revizuirii.
Provocările armonizării: Diferențe între MDR UE, FDA și alte piețe
Cerințele regionale diferite creează o complexitate în ceea ce privește conformitatea:
- MDR UE impune evaluări clinice pentru marcarea CE
- FDA pune accent pe echivalența bazată pe precedent
- PMDA Japoniei necesită conformitatea cu ISO 80369-6 pentru conexiunile neuraxiale
- ANVISA din Brazilia impune testarea locală a biocompatibilității
Aceste discrepanțe obligă producătorii să mențină 2–3 variante de dispozitive, ceea ce crește costurile de dezvoltare cu 500.000–1,2 milioane USD pe produs (Raportul Global privind Conformitatea MedTech 2024).
Tendință: Strictețe sporită în cerințele de supraveghere post-comercializare
Reglementatorii solicită acum date privind performanța în condiții reale, cerințele FDA pentru studiile post-comercializare crescând cu 40% din 2021. Producătorii trebuie să implementeze sisteme electronice de urmărire pentru a raporta evenimentele adverse în termen de 15 zile—cu 50% mai rapid decât termenele din 2020. Ratele de eșec la auditurile de supraveghere au crescut până la 22% la nivelul întregii industrii, subliniind necesitatea unei gestionări proactive a riscurilor (Revista Medical Device Regulatory 2023).
Certificări ale managementului calității și al producției pentru furnizorii de traductoare IBP
Importanța ISO 13485 în proiectarea și producția traductoarelor IBP
Certificarea ISO 13485 este foarte importantă pentru managementul calității în rândul furnizorilor de traductoare IBP. Ajută la menținerea controlului pe toate etapele, de la proiectare până la producție și chiar după ce produsele ajung pe piață. Ceea ce o diferențiază de standardele obișnuite de calitate este modul în care abordează cerințele specifice ale dispozitivelor medicale. Gândiți-vă la aspecte precum posibilitatea de a urmări proiectele până la originea lor, asigurarea faptului că sterilizarea funcționează corespunzător și luarea deciziilor pe baza riscurilor reale, nu a presupunerilor. Analiza datelor din 2023 privind dispozitivele medicale retrase de pe piață arată și un lucru interesant: companiile cu certificare ISO 13485 au avut aproximativ jumătate din numărul problemelor legate de neconformitate comparativ cu cele fără această certificare. Atunci când vorbim despre traductoare IBP în mod specific, urmarea acestor directive înseamnă că senzorii de presiune și căile pentru fluide rămân în limite stricte de siguranță și funcționează fiabil pe întreaga durată de viață în spitale și clinici.
Conformitatea cu Practicile de Producție Bună (GMP) în producția contractuală
Urmarea recomandărilor privind bunele practici de fabricare (GMP) ajută producătorii contractanți să mențină un nivel de consistență în fabricarea transductorilor IBP care să respecte atât standardele FDA, cât și cele ale reglementării UE MDR. Ce face ca GMP să fie atât de important? Ei bine, acoperă trei domenii principale: păstrarea corectă a înregistrărilor, asigurarea calibrării corespunzătoare a echipamentelor și instruirea adecvată a personalului. Aceste aspecte au un impact direct asupra preciziei măsurătorilor realizate de acești monitori invazivi ai tensiunii arteriale. Analizând datele recente din inspecții, unitățile complet conforme cu GMP obțin o rată de succes de aproximativ 98% la prima încercare în testele de sterilitate. Acest rezultat este mult mai bun decât cele aproximativ 72% observate în locurile care urmează parțial aceste reguli. Atunci când companiile externalizează părți ale procesului de fabricație, cum ar fi crearea diafragmelor transductorului sau asamblarea acestora, respectarea GMP devine și mai esențială. Mici erori în procedurile de lucru în sală curată de la aceste site-uri terțe pot afecta grav calitatea produsului final.
Întrebări frecvente (FAQ)
Ce anume face un transductor IBP?
Transductorii IBP convertesc citirile de presiune din artere în semnale electrice, permițând monitorizarea în timp real a tensiunii arteriale în cadrul procedurilor medicale critice.
De ce este importantă certificarea CE pentru transductorii IBP?
Certificarea CE asigură faptul că transductorii IBP respectă standardele stricte de siguranță necesare pentru dispozitivele medicale vândute în Europa, garantând siguranța pacientului.
Cum ajută standardele ISO 10993 la fabricarea transductorilor IBP?
Standardele ISO 10993 permit producătorilor să evalueze materialele din punct de vedere al biocompatibilității, minimizând reacțiile biologice adverse cauzate de contactul cu pacientul.
Care este rolul Organismelor Notificate în conformitatea CE?
Organismele Notificate acționează ca auditori independenți, validând faptul că transductorii IBP respectă cerințele regulatorii înainte de intrarea pe piață.
De ce este importantă norma ISO 13485 pentru managementul calității în producția transductorilor IBP?
ISO 13485 asigură faptul că furnizorii de dispozitive medicale mențin standarde înalte în toate etapele, de la proiectare și producție până la practicile post-vânzare, pentru a garanta calitatea și siguranța produsului.
Cuprins
- Înțelegerea transductoarelor IBP și importanța lor clinică
-
Certificare CE și conformitate cu reglementarea UE MDR pentru transductori IBP
- Ce înseamnă certificarea CE pentru siguranța și performanța dispozitivelor medicale
- Pași pentru obținerea marcării CE în conformitate cu MDR UE pentru traductoarele IBP
- Rolul Organismelor Notificate în validarea conformității CE
- Autocertificare vs. Evaluare de către terți pentru dispozitivele din clasa IIa
-
Standardele ISO 10993 privind biocompatibilitatea și ISO 14971 privind managementul riscurilor
- De ce este importantă norma ISO 10993 pentru componentele care vin în contact cu pacientul în sistemele IBP
- Evaluarea riscurilor de citotoxicitate, sensibilizare și iritație
- Studiu de caz: Eșecuri în selecția materialelor care au condus la neconformitate
- Integrarea testării biocompatibilității cu managementul riscurilor conform ISO 14971
- Aprobarea FDA și cerințele reglementare globale pentru traductoarele IBP
- Certificări ale managementului calității și al producției pentru furnizorii de traductoare IBP
-
Întrebări frecvente (FAQ)
- Ce anume face un transductor IBP?
- De ce este importantă certificarea CE pentru transductorii IBP?
- Cum ajută standardele ISO 10993 la fabricarea transductorilor IBP?
- Care este rolul Organismelor Notificate în conformitatea CE?
- De ce este importantă norma ISO 13485 pentru managementul calității în producția transductorilor IBP?