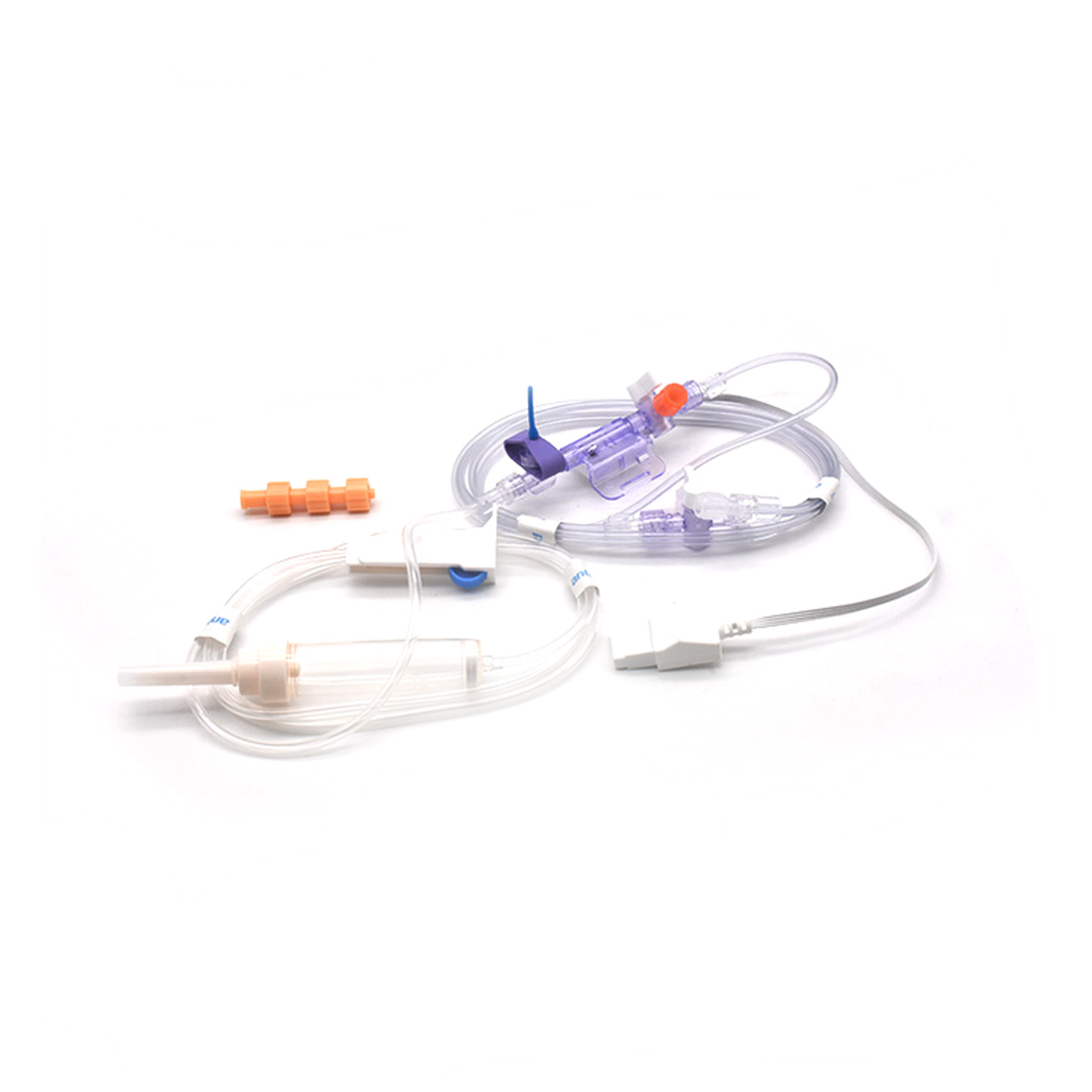
Entendendo os Transdutores IBP e sua Importância Clínica
O que é um transdutor IBP e como ele funciona?
Os transdutores de pressão arterial invasiva (IBP) são dispositivos médicos que obtêm leituras reais da pressão nas artérias conectadas por meio de cateteres e as convertem em sinais elétricos para que os médicos possam acompanhar em tempo real o que está acontecendo. Esses dispositivos normalmente possuem sensores mantidos estéreis, capazes de detectar até as menores variações nos níveis de pressão, enviando em seguida todas essas informações aos monitores por meio de sistemas preenchidos com fluido. Os modelos mais recentes atingem uma precisão de cerca de ±2 mmHg, o que é extremamente importante para detectar quedas ou picos perigosos na pressão arterial durante cirurgias. Uma pesquisa publicada no ano passado no Journal of Critical Care Medicine revelou algo interessante também – pacientes monitorados com esses transdutores precisos tiveram seus episódios de pressão baixa identificados 42% mais rapidamente do que com técnicas não invasivas convencionais. Esse tipo de aviso precoce faz grande diferença nos centros cirúrgicos, onde cada segundo conta.
Importância Clínica do Monitoramento Preciso da Pressão Arterial Invasiva
Para pacientes que necessitam de vasoconstritores ou ajuda para estabilizar sua circulação, a monitorização invasiva da pressão arterial (IBP) ainda é considerada a melhor abordagem. Os métodos oscilométricos simplesmente não são suficientes quando precisamos de leituras batimento a batimento, tão importantes em casos de choque séptico, após acidentes graves ou durante cirurgias cardíacas. Os médicos dependem da rapidez com que esses monitores reagem, normalmente em menos de um segundo, para decidir sobre questões como a administração de fluidos. A experiência clínica tem mostrado que mesmo esperar cinco minutos inteiros para corrigir problemas na pressão arterial pode levar a uma taxa de mortalidade cerca de 15% maior. Um estudo recente sobre práticas em UTIs revelou também algo interessante: hospitais que implementaram configurações padronizadas de transdutores IBP registraram aproximadamente 31% menos erros ao ajustar medicamentos em suas unidades de terapia intensiva.
Certificação CE e Conformidade com o Regulamento Europeu de Dispositivos Médicos (EU MDR) para Transdutores IBP
O que a Certificação CE Significa para a Segurança e Desempenho de Dispositivos Médicos
Obter a certificação CE de acordo com o Regulamento da União Europeia sobre Dispositivos Médicos (MDR) significa basicamente que um transdutor IBP passa por rigorosos testes de segurança necessários para ser comercializado na Europa. A marcação CE indica que o produto segue normas importantes, como a IEC 60601 para equipamentos eletrônicos médicos, e também atende às regras de gestão de riscos da ISO 14971. Para dispositivos que cumprem as regulamentações MDR, existe todo um processo de testagem da compatibilidade segura dos materiais que entram em contato com pacientes, garantindo que não causem danos celulares ou irritações. A maioria dos dispositivos de monitoramento da pressão arterial se enquadra na categoria Classe IIa ou superior, o que significa que os fabricantes não podem simplesmente declarar conformidade por conta própria, mas precisam de especialistas independentes para verificar se tudo funciona corretamente antes de chegar ao mercado.
Etapas para obter a Marcação CE segundo o MDR da UE para transdutores IBP
- Classificar o dispositivo : Determinar a classificação de risco (normalmente Classe IIa para transdutores IBP) conforme o Anexo VIII do MDR.
- Implementar um sistema de gestão da qualidade (QMS) : Estabeleça um sistema de gestão da qualidade certificado conforme a ISO 13485.
- Prepare a documentação técnica : Compile especificações de design, avaliações de risco e relatórios de avaliação clínica.
- Contrate um Organismo Notificado : Para dispositivos da Classe IIa+, submeta-se a auditorias dos arquivos técnicos e processos de fabricação.
- Afixe a marcação CE : Após a avaliação de conformidade, registre o dispositivo no EUDAMED. Oitenta e nove por cento dos atrasos decorrem de avaliações clínicas incompletas ou documentação do SGQ desatualizada, segundo a SimplerQMS.
Papel dos Organismos Notificados na validação da conformidade CE
Os Organismos Notificados, ou ONs abreviadamente, atuam como auditores independentes no que diz respeito a dispositivos médicos classificados como de maior risco. A sua função envolve analisar toda a documentação técnica e realizar visitas in loco aos locais de fabrico para inspeções. Ao analisar especificamente os transdutores IBP, estes organismos verificam se tudo está em conformidade com os requisitos descritos no Anexo II do Regulamento de Dispositivos Médicos (MDR). Isso inclui garantir que as versões digitais possuam validação adequada de software e que os produtos descartáveis cumpram rigorosos padrões de esterilidade. De acordo com uma pesquisa recente de 2023, a maioria dos problemas identificados pelos ONs decorre de duas áreas principais: empresas que não possuem estratégias sólidas de vigilância pós-comercialização ou que não fornecem dados suficientes de biocompatibilidade. Essas constatações destacam os pontos nos quais os fabricantes precisam concentrar suas atenções para obter sucesso nas auditorias.
Auto-certificação versus Avaliação por Terceiros para Dispositivos da Classe IIa
Embora dispositivos não estéreis da Classe I possam ser autodeclarados, os transdutores de PIA da Classe IIa exigem supervisão obrigatória de um Organismo Notificado. A auto-certificação aplica-se apenas a acessórios de baixo risco, como cabos reutilizáveis; componentes sensores de pressão exigem revisão completa por terceiros. Atualizações recentes do MDR exigem auditorias anuais do Organismo Notificado para fabricantes de Classe IIa, reduzindo em 34% as entradas ilegais no mercado desde 2021.
Normas de Biocompatibilidade ISO 10993 e de Gestão de Riscos ISO 14971
Por que a ISO 10993 é importante para componentes em contato com o paciente em sistemas de PIA
Para transdutores de PIA que realmente tocam os pacientes, seguir as normas ISO 10993 é praticamente obrigatório se quisermos evitar aquelas reações biológicas indesejadas. O objetivo principal dessa norma é determinar o que acontece quando as pessoas são expostas por longos períodos a todo tipo de materiais, como peças plásticas, colas usadas na montagem e diversos componentes sensores. De acordo com pesquisas publicadas no ano passado, cerca de um em cada cinco casos de irritação cutânea relatados em hospitais decorreram de dispositivos médicos fabricados com materiais que não atendiam aos requisitos da ISO 10993-10. As empresas que incorporam esses critérios em seus processos produtivos por meio de Planos de Avaliação Biológica adequados tendem a enfrentar menos problemas, pois testam como diferentes materiais reagem ao entrar em contato com fluidos e tecidos corporais ao longo do tempo.
Avaliação dos Riscos de Citotoxicidade, Sensibilização e Irritação
A ISO 10993 exige três testes principais para os materiais dos transdutores de PIA:
- Citotoxicidade (ISO 10993-5): Avalia a viabilidade celular após exposição ao material (morte de até 70% das células permitida)
- Sensibilização (ISO 10993-10): Avalia o potencial de resposta alérgica utilizando testes de maximização em cobaias
- Irritação (ISO 10993-10): Avalia os riscos de inflamação localizada por meio de testes com curativos epidérmicos
Dispositivos que falham em qualquer teste enfrentam taxas de recolhimento pela FDA 89% maiores em comparação com alternativas totalmente conformes (Relato de Dispositivos Médicos, 2022).
Estudo de Caso: Falhas na Seleção de Materiais que Levaram à Não Conformidade
Uma auditoria da UE MDR de 2021 revelou que 32% dos transdutores IBP rejeitados utilizavam tubos de silicone sem certificação ISO 10993-33 para contato sanguíneo prolongado. O recolhimento de US$ 2,7 milhões realizado pelo fabricante resultou de:
- Assumir que materiais "de grau médico" atendiam automaticamente aos padrões de biocompatibilidade
- Omitir testes de envelhecimento acelerado para riscos de lixiviação química
- Documentação inadequada dos impactos da esterilização na segurança do material
Integração dos Testes de Biocompatibilidade com a Gestão de Riscos conforme ISO 14971
A combinação dos dados da ISO 10993 com o framework de risco da ISO 14971 permite que os fabricantes:
- Quantifiquem riscos biológicos utilizando matrizes de gravidade/probabilidade de perigo
- Implementem controles como substituições de materiais ou revestimentos biocompatíveis
- Monitorem eventos adversos pós-comercialização por meio de processos alinhados à ISO 13485
Essa abordagem dupla reduz mudanças de design em estágios avançados em 41% em comparação com testes de biocompatibilidade isolados (MedTech Quality Benchmark, 2023).
Liberação pela FDA e Requisitos Regulatórios Globais para Transdutores IBP
Processo de Liberação FDA 510(k) e Equivalência Substancial
Para os fabricantes de dispositivos médicos, obter a liberação pelo programa 510(k) da FDA significa demonstrar que seu produto é substancialmente equivalente a algo já disponível no mercado. Cerca de três quartos de todos os dispositivos foram aprovados dessa forma em 2023, segundo dados do setor. Cumprir esses requisitos exige um trabalho sério de testes e rigorosa aderência às regras do 21 CFR Parte 820 sobre sistemas de qualidade. Empresas que possuem certificação ISO 13485 tendem a ter seus pedidos processados cerca de 30 por cento mais rapidamente na FDA. Isso parece resultar de práticas de organização de documentação e arquivamento mais eficientes, alinhadas ao que os reguladores esperam ver durante a análise.
Desafios de Harmonização: Diferenças entre o Regulamento UE MDR, a FDA e outros mercados
Requisitos regionais divergentes criam complexidade de conformidade:
- O Regulamento UE MDR exige avaliações clínicas para a marcação CE
- A FDA enfatiza a equivalência baseada em dispositivo antecedente
- A PMDA do Japão exige conformidade com a norma ISO 80369-6 para conexões neuraxiais
- A ANVISA do Brasil exige testes locais de biocompatibilidade
Essas discrepâncias obrigam os fabricantes a manterem 2–3 variantes do dispositivo, aumentando os custos de desenvolvimento em US$ 500 mil–US$ 1,2 milhão por produto (Relatório Global de Conformidade Médica 2024).
Tendência: Aumento da rigorosidade nos requisitos de vigilância pós-comercialização
Os reguladores agora exigem dados de desempenho no mundo real, com um aumento de 40% nos requisitos de estudos pós-comercialização pela FDA desde 2021. Os fabricantes devem implementar sistemas eletrônicos de rastreabilidade para relatar eventos adversos em até 15 dias — 50% mais rápido que os prazos de 2020. As taxas de falha em auditorias de vigilância aumentaram para 22% em toda a indústria, destacando a necessidade de gestão proativa de riscos (Revista de Regulamentação de Dispositivos Médicos 2023).
Gestão da Qualidade e Certificações de Fabricação para Fornecedores de Transdutores IBP
Importância da ISO 13485 no Projeto e Produção de Transdutores IBP
A certificação ISO 13485 é realmente importante para o gerenciamento da qualidade entre os fornecedores de transdutores IBP. Ela ajuda a manter o controle em todas as etapas, desde o projeto até a produção e também após os produtos chegarem ao mercado. O que diferencia isso dos padrões de qualidade comuns é a forma como aborda as necessidades específicas dos dispositivos médicos. Pense em aspectos como a capacidade de rastrear os projetos até suas origens, garantir que a esterilização funcione corretamente e tomar decisões com base em riscos reais, e não em suposições. Analisar dados de 2023 sobre dispositivos médicos recolhidos revela algo interessante também. Empresas com certificação ISO 13485 tiveram aproximadamente metade dos problemas em atender aos padrões em comparação às que não possuíam essa certificação. Quando falamos especificamente de transdutores IBP, seguir essas diretrizes significa que sensores de pressão e caminhos de fluidos permanecem dentro das margens rigorosas de segurança e apresentam desempenho confiável durante todo o seu ciclo de vida em hospitais e clínicas.
Alinhamento com as Boas Práticas de Fabricação (GMP) na Fabricação por Contrato
Seguir as diretrizes da Boa Prática de Fabricação (GMP) ajuda os fabricantes terceirizados a manterem a consistência na produção de transdutores IBP que atendam tanto às normas da FDA quanto às da EU MDR. O que torna a GMP tão importante? Ela abrange três áreas principais: manter registros adequados, garantir a calibração correta dos equipamentos e capacitar adequadamente a equipe. Esses aspectos têm um impacto direto na precisão das medições dos monitores de pressão sanguínea invasiva. Analisando dados recentes de auditorias, instalações totalmente conformes com a GMP obtêm cerca de 98% de sucesso na primeira tentativa em testes de esterilidade. Isso é muito melhor do que os aproximadamente 72% observados em locais que seguem parcialmente essas regras. Quando empresas terceirizam partes do processo de fabricação, como a criação das diafragmas dos transdutores ou sua montagem, aderir à GMP torna-se ainda mais essencial. Pequenos erros nos procedimentos de sala limpa nesses fornecedores podem comprometer seriamente a qualidade do produto final.
Perguntas Frequentes (FAQ)
O que exatamente um transdutor IBP faz?
Os transdutores IBP convertem leituras de pressão das artérias em sinais elétricos, permitindo o monitoramento em tempo real da pressão arterial durante procedimentos médicos críticos.
Por que a certificação CE é importante para os transdutores IBP?
A certificação CE garante que os transdutores IBP atendam a rigorosas normas de segurança exigidas para dispositivos médicos vendidos na Europa, assegurando a segurança do paciente.
Como as normas ISO 10993 auxiliam na fabricação de transdutores IBP?
As normas ISO 10993 permitem aos fabricantes avaliar a biocompatibilidade dos materiais, minimizando reações biológicas adversas decorrentes do contato com o paciente.
Qual é o papel dos Organismos Notificados na conformidade com a CE?
Os Organismos Notificados atuam como auditores independentes, validando que os transdutores IBP atendam aos requisitos regulamentares antes de ingressarem no mercado.
Por que a ISO 13485 é importante para a gestão da qualidade na produção de transdutores IBP?
A ISO 13485 garante que os fornecedores de dispositivos médicos mantenham altos padrões em todo o ciclo de desenvolvimento, produção e práticas pós-comercialização, assegurando a qualidade e segurança do produto.
Sumário
- Entendendo os Transdutores IBP e sua Importância Clínica
-
Certificação CE e Conformidade com o Regulamento Europeu de Dispositivos Médicos (EU MDR) para Transdutores IBP
- O que a Certificação CE Significa para a Segurança e Desempenho de Dispositivos Médicos
- Etapas para obter a Marcação CE segundo o MDR da UE para transdutores IBP
- Papel dos Organismos Notificados na validação da conformidade CE
- Auto-certificação versus Avaliação por Terceiros para Dispositivos da Classe IIa
-
Normas de Biocompatibilidade ISO 10993 e de Gestão de Riscos ISO 14971
- Por que a ISO 10993 é importante para componentes em contato com o paciente em sistemas de PIA
- Avaliação dos Riscos de Citotoxicidade, Sensibilização e Irritação
- Estudo de Caso: Falhas na Seleção de Materiais que Levaram à Não Conformidade
- Integração dos Testes de Biocompatibilidade com a Gestão de Riscos conforme ISO 14971
- Liberação pela FDA e Requisitos Regulatórios Globais para Transdutores IBP
- Gestão da Qualidade e Certificações de Fabricação para Fornecedores de Transdutores IBP
-
Perguntas Frequentes (FAQ)
- O que exatamente um transdutor IBP faz?
- Por que a certificação CE é importante para os transdutores IBP?
- Como as normas ISO 10993 auxiliam na fabricação de transdutores IBP?
- Qual é o papel dos Organismos Notificados na conformidade com a CE?
- Por que a ISO 13485 é importante para a gestão da qualidade na produção de transdutores IBP?