Zrozumienie przetworników IBP i ich znaczenia klinicznego
Co to jest przetwornik IBP i jak działa?
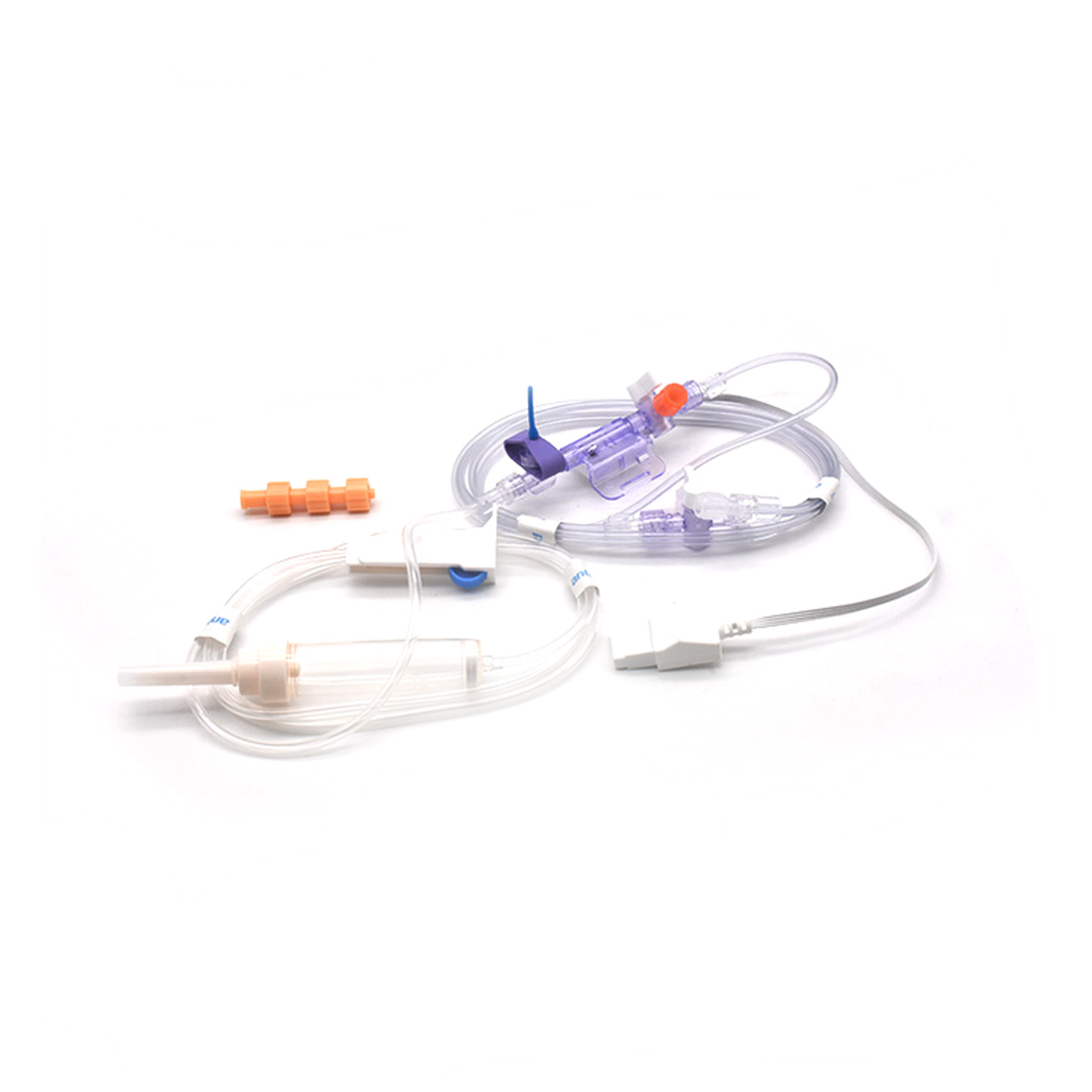
Przetworniki inwazyjnego ciśnienia krwi (IBP) to urządzenia medyczne, które odczytują rzeczywiste wartości ciśnienia z tętnic połączonych za pomocą cewników i przekształcają je na sygnały elektryczne, umożliwiając lekarzom obserwację bieżących zmian w czasie rzeczywistym. Urządzenia te posiadają zazwyczaj sterylnie przygotowane czujniki, które reagują nawet na najmniejsze wahania poziomu ciśnienia, a następnie przesyłają zebrane informacje do monitorów poprzez układy wypełnione cieczą. Nowsze modele osiągają dokładność rzędu plus minus 2 mmHg, co ma ogromne znaczenie podczas wykrywania niebezpiecznych spadków lub skoków ciśnienia krwi w trakcie operacji. Badania opublikowane w zeszłym roku w Journal of Critical Care Medicine wykazały również interesujący fakt – u pacjentów monitorowanych tymi precyzyjnymi przetwornikami przypadki niskiego ciśnienia były wykrywane o 42% szybciej niż przy użyciu standardowych metod nieinwazyjnych. Tego rodzaju wczesne ostrzeżenie ma ogromne znaczenie w salach operacyjnych, gdzie każda sekunda się liczy.
Znaczenie kliniczne dokładnego inwazyjnego monitorowania ciśnienia krwi
U pacjentów potrzebujących środków nacisku na naczynia krwionośne lub pomagających ustabilizować krążenie, inwazyjne monitorowanie ciśnienia krwi (IBP) jest nadal uważane za najlepsze podejście. Metody oscylometryczne nie wystarczą, gdy potrzebujemy odczytu bicia po biciu, który jest tak ważny w przypadku szoku septycznego, po poważnym wypadku lub operacji serca. Lekarze zależą od szybkości reakcji tych monitorów, zazwyczaj poniżej jednej sekundy, aby podjąć decyzję o podawaniu płynów. Z doświadczenia klinicznego wynika, że nawet pięć minut oczekiwania na poprawienie ciśnienia krwi może prowadzić do o 15% większej śmiertelności. Przyjrzenie się praktykom w intensywnej terapii wykazało niedawno coś interesującego: szpitale, które wdrożyły standardowe ustawienia przetwornika IBP, odnotowały o około 31% mniej błędów podczas dostosowywania leków na oddziałach intensywnej terapii.
Certyfikacja CE i zgodność z MDR UE dla przetworników IBP
Co oznacza certyfikacja CE dla bezpieczeństwa i wydajności wyrobów medycznych
Uzyskanie certyfikatu CE zgodnie z rozporządzeniem Unii Europejskiej w sprawie wyrobów medycznych (MDR) oznacza przede wszystkim, że przetwornik IBP przechodzi rygorystyczne kontrole bezpieczeństwa wymagane do sprzedaży w Europie. Oznakowanie CE wskazuje, że produkt spełnia ważne normy, takie jak IEC 60601 dotyczące sprzętu elektronicznego medycznego, oraz przestrzega zasad zarządzania ryzykiem określonych w ISO 14971. W przypadku urządzeń zgodnych z przepisami MDR przewidziano kompleksowy proces testowania biokompatybilności materiałów stykających się z pacjentem, aby upewnić się, że nie spowodują uszkodzeń komórkowych ani podrażnień. Większość urządzeń do pomiaru ciśnienia krwi należy do kategorii IIa lub wyższej, co oznacza, że producenci nie mogą samodzielnie deklarować zgodności, lecz muszą powołać niezależnych ekspertów, którzy zweryfikują poprawność działania przed wprowadzeniem produktu na rynek.
Kroki prowadzące do uzyskania oznakowania CE zgodnie z unijnym rozporządzeniem MDR dla przetworników IBP
- Skategoryzuj urządzenie : Określ klasę ryzyka (zwykle klasa IIa dla przetworników IBP) zgodnie z załącznikiem VIII rozporządzenia MDR.
- Wdrożenie systemu zarządzania jakością (QMS) : Ustanów system zarządzania jakością certyfikowany zgodnie z normą ISO 13485.
- Przygotuj dokumentację techniczną : Zcompiluj specyfikacje projektowe, oceny ryzyka oraz raporty oceny klinicznej.
- Skontaktuj się z Notyfikowanym Organem : W przypadku urządzeń klasy IIa i wyższych, przejdź audyty plików technicznych i procesów produkcyjnych.
- Umieść znak CE : Po ocenie zgodności zarejestruj urządzenie w EUDAMED. Zgodnie z danymi SimplerQMS, 89 procent opóźnień wynika z niekompletnych ocen klinicznych lub przestarzałej dokumentacji systemu zarządzania jakością.
Rola Notyfikowanych Organów w weryfikacji zgodności z wymogami CE
Organy notyfikowane, zwane również NB, działają jako niezależni audytorzy w przypadku urządzeń medycznych sklasyfikowanych jako wyższego ryzyka. Ich zadanie obejmuje analizę wszystkich dokumentów technicznych oraz fizyczne wizyty na terenie zakładów produkcyjnych w celu przeprowadzenia inspekcji. W odniesieniu do przetworników IBP, organy te sprawdzają, czy wszystko jest zgodne z wymaganiami określonymi w Załączniku II rozporządzenia MDR. Obejmuje to zapewnienie odpowiedniej walidacji oprogramowania dla wersji cyfrowych oraz spełnienie ścisłych standardów sterylności przez elementy jednorazowe. Zgodnie z najnowszym badaniem przeprowadzonym w 2023 roku, większość problemów wykrytych przez organy notyfikowane wynika z dwóch głównych obszarów: braku u producentów solidnych strategii nadzoru po wprowadzeniu na rynek lub niewystarczających danych dotyczących biokompatybilności. Te ustalenia wskazują, na które obszary producenci powinni skupić uwagę, aby pomyślnie przejść audyty.
Samodzielna certyfikacja a ocena przez podmiot trzeci dla urządzeń klasy IIa
Chociaż urządzenia niebieskie klasy I mogą być samocertyfikowane, przetworniki IBP klasy IIa wymagają obowiązkowego nadzoru organu notyfikowanego. Samocertyfikacja dotyczy wyłącznie niskiego ryzyka akcesoriów, takich jak wielokrotnego użytku kable; komponenty pomiaru ciśnienia wymagają pełnej oceny przez podmiot trzeci. Najnowsze aktualizacje MDR wymagają corocznych audytów ON od producentów klasy IIa, co zmniejszyło liczbę niezgodnych wprowadzeń na rynek o 34% od 2021 roku.
Standardy biokompatybilności ISO 10993 i zarządzania ryzykiem ISO 14971
Dlaczego norma ISO 10993 jest ważna dla komponentów wchodzących w kontakt z pacjentem w systemach IBP
W przypadku przetworników IBP, które faktycznie stykają się z pacjentami, przestrzeganie norm ISO 10993 jest niemal obowiązkowe, jeśli chcemy uniknąć nieprzyjemnych reakcji biologicznych. Cały sens tej normy polega na ocenie, co się dzieje, gdy ludzie są narażeni przez dłuższy czas na różnorodne materiały, takie jak elementy plastikowe, kleje stosowane w procesie montażu czy różne czujniki. Zgodnie z badaniami opublikowanymi w zeszłym roku, około jedna na pięć przypadków podrażnień skóry zgłoszonych w szpitalach pochodziła z urządzeń medycznych wykonanych z materiałów, które nie spełniały wymagań normy ISO 10993-10. Firmy, które integrują te procedury w swoim procesie produkcyjnym poprzez odpowiednie Plany Oceny Biologicznej, napotykają mniej problemów, ponieważ testują, w jaki sposób różne materiały reagują przy kontakcie z rzeczywistymi płynami ustrojowymi i tkankami w czasie.
Ocena ryzyka cytotoksyczności, uczulenia i podrażnień
ISO 10993 wymaga trzech podstawowych testów dla materiałów przetworników IBP:
- Cytotoksyczność (ISO 10993-5): Mierzy żywotność komórek po ekspozycji na materiał (dozwolone jest uszkodzenie 70% komórek)
- Sensytyzacja (ISO 10993-10): Ocenia potencjał reakcji alergicznej za pomocą testów maksymalizacji u świnki morskiej
- Podrażnienie (ISO 10993-10): Ocenia ryzyko lokalnego zapalenia poprzez test przylepów epidermalnych
Urządzenia, które nie przejdą któregoś z testów, są narażone na o 89% wyższe ryzyko wycofania przez FDA w porównaniu z alternatywami pełniące wszystkie wymagania (Medical Device Reporting, 2022).
Studium przypadku: Błędy w doborze materiałów prowadzące do braku zgodności
Audyt UE MDR z 2021 roku ujawnił, że 32% odrzuconych przetworników IBP wykorzystywało rurki silikonowe bez certyfikatu ISO 10993-33 na długotrwały kontakt z krwią. Wycofanie produktu przez producenta za 2,7 mln USD wynikło z:
- Zakładania, że materiały „medyczne” automatycznie spełniają normy biokompatybilności
- Pominięcia testów przyspieszonego starzenia pod kątem ryzyka wyciekania substancji chemicznych
- Niewystarczająca dokumentacja wpływu sterylizacji na bezpieczeństwo materiału
Integrowanie badań biokompatybilności z zarządzaniem ryzykiem zgodnie z normą ISO 14971
Łączenie danych z normy ISO 10993 z ramami ryzyka według ISO 14971 pozwala producentom na:
- Ilościowe określanie ryzyk biologicznych za pomocą macierzy ciężkości zagrożeń/prawdopodobieństwa
- Wdrażanie środków zapobiegawczych, takich jak wymiana materiałów lub stosowanie biokompatybilnych powłok
- Monitorowanie niepożądanych zdarzeń pogwarancyjnych poprzez procesy zgodne z normą ISO 13485
Takie podejście dwustopniowe zmniejsza liczbę późnych zmian konstrukcyjnych o 41% w porównaniu z samodzielnym testowaniem biokompatybilności (MedTech Quality Benchmark, 2023).
Zezwolenie FDA i globalne wymagania regulacyjne dla przetworników IBP
Proces uzyskiwania zezwolenia FDA 510(k) i istotna równoważność
Dla producentów wyrobów medycznych uzyskanie zezwolenia w ramach programu FDA 510(k) oznacza wykazanie, że ich produkt jest istotnie równoważny temu już znajdującemu się na rynku. Według danych branżowych około trzech czwartych wszystkich urządzeń zostało zatwierdzonych właśnie tą drogą w 2023 roku. Spełnienie tych wymagań wiąże się z poważnymi pracami badawczymi oraz rygorystycznym przestrzeganiem przepisów 21 CFR część 820 dotyczących systemów jakości. Firmy posiadające certyfikat ISO 13485 zazwyczaj doświadczają szybszego procesu przeglądu wniosków przez FDA — średnio o około 30 procent krótszego. Wydaje się, że wynika to z lepiej zorganizowanej dokumentacji i praktyk archiwizacyjnych, które odpowiadają oczekiwaniom regulatorów podczas oceny.
Wyzwania harmonizacji: różnice między unijnym rozporządzeniem MDR, FDA a innymi rynkami
Rozbieżne wymagania regionalne tworzą złożoność w zakresie zgodności:
- Unijne rozporządzenie MDR wymaga ocen klinicznych dla oznakowania CE
- FDA podkreśla równoważność opartą na poprzedniku
- Japońskie PMDA wymaga zgodności z normą ISO 80369-6 w odniesieniu do połączeń neuraksjalnych
- Brazylijska ANVISA wymusza lokalne testy biokompatybilności
Te rozbieżności zmuszają producentów do utrzymywania 2–3 wariantów urządzeń, co zwiększa koszty rozwoju o 500 tys. – 1,2 mln USD na produkt (Global MedTech Compliance Report 2024).
Trend: Wzrost rygoru wymagań dotyczących nadzoru poprzez rynkowy
Regulatory obecnie wymagają danych o rzeczywistej wydajności działania, a liczba badań post-marketowych wymaganych przez FDA wzrosła o 40% od 2021 roku. Producenci muszą wdrożyć elektroniczne systemy śledzenia, aby zgłaszać niepożądane zdarzenia w ciągu 15 dni – o 50% szybciej niż terminy z 2020 roku. Stopy błędów podczas audytów nadzorczych wzrosły do 22% w skali branży, co podkreśla konieczność proaktywnego zarządzania ryzykiem (Medical Device Regulatory Journal 2023).
Zarządzanie jakością i certyfikaty produkcyjne dla dostawców przetworników IBP
Znaczenie normy ISO 13485 w projektowaniu i produkcji przetworników IBP
Certyfikat ISO 13485 ma ogromne znaczenie dla zarządzania jakością wśród dostawców przetworników IBP. Pomaga on utrzymać kontrolę na wszystkich etapach — od projektowania, przez produkcję, aż po to, co dzieje się po wprowadzeniu produktów na rynek. To, co odróżnia ten standard od typowych norm jakości, to sposób, w jaki uwzględnia on specyficzne wymagania urządzeń medycznych. Chodzi tu na przykład o możliwość śledzenia projektów do ich źródeł, zapewnienie skuteczności procesów sterylizacji oraz podejmowanie decyzji opartych na rzeczywistych ryzykach, a nie domysłach. Analiza danych z 2023 roku dotyczących wycofanych urządzeń medycznych ujawnia ciekawy fakt: firmy posiadające certyfikat ISO 13485 miały około dwa razy mniej problemów z zgodnością niż te bez certyfikatu. W przypadku przetworników IBP przestrzeganie tych wytycznych oznacza, że czujniki ciśnienia i ścieżki przepływu płynów pozostają w ściśle określonych granicach bezpieczeństwa i działają niezawodnie przez cały okres użytkowania w szpitalach i klinikach.
Zgodność z dobrą praktyką wytwarzania (GMP) w produkcji kontraktowej
Przestrzeganie wytycznych zasad dobrej praktyki produkcyjnej (GMP) pomaga producentom kontraktowym utrzymać spójność podczas wytwarzania przetworników IBP spełniających normy FDA i unijnego rozporządzenia MDR. Dlaczego GMP jest tak ważne? Otóż obejmuje trzy główne obszary: prowadzenie odpowiedniej dokumentacji, zapewnienie kalibracji sprzętu oraz właściwe szkolenie personelu. Te aspekty mają bezpośredni wpływ na dokładność pomiarów uzyskiwanych za pomocą monitorów inwazyjnego ciśnienia krwi. Analiza najnowszych danych z audytów wykazuje, że zakładom całkowicie zgodnym z GMP udaje się osiągnąć około 98% skuteczności przy pierwszej próbie testów sterylności. To znacznie lepszy wynik niż około 72% notowanych w miejscach jedynie częściowo przestrzegających tych zasad. Gdy firmy outsource’ują części procesu produkcyjnego, takie jak tworzenie membran przetworników lub ich montaż, przestrzeganie GMP staje się jeszcze ważniejsze. Niewielkie błędy w procedurach czystych pomieszczeń w tych firmach trzecich mogą poważnie wpłynąć na jakość końcowego produktu.
Często Zadawane Pytania (FAQ)
Do czego służy dokładnie przetwornik IBP?
Transducery IBP przekształcają odczyty ciśnienia z tętnic na sygnały elektryczne, umożliwiając monitorowanie ciśnienia krwi w czasie rzeczywistym podczas krytycznych procedur medycznych.
Dlaczego certyfikat CE jest ważny dla transducerów IBP?
Certyfikat CE gwarantuje, że transducery IBP spełniają rygorystyczne wymagania bezpieczeństwa obowiązujące dla urządzeń medycznych sprzedawanych w Europie, zapewniając bezpieczeństwo pacjentów.
Jak normy ISO 10993 pomagają w produkcji transducerów IBP?
Normy ISO 10993 pozwalają producentom oceniać materiały pod kątem biokompatybilności, minimalizując niepożądane reakcje biologiczne wynikające ze styku z pacjentem.
Jaką rolę odgrywają upoważnione organy w zakresie zgodności z dyrektywą CE?
Upoważnione organy działają jako niezależni audytorzy, weryfikując, czy transducery IBP spełniają wymagania regulacyjne przed wprowadzeniem ich na rynek.
Dlaczego norma ISO 13485 jest ważna dla zarządzania jakością w produkcji transducerów IBP?
ISO 13485 zapewnia, że dostawcy urządzeń medycznych utrzymują wysokie standardy w zakresie projektowania, produkcji i działań po wprowadzeniu produktu na rynek, aby zagwarantować jakość i bezpieczeństwo produktów.
Spis treści
- Zrozumienie przetworników IBP i ich znaczenia klinicznego
-
Certyfikacja CE i zgodność z MDR UE dla przetworników IBP
- Co oznacza certyfikacja CE dla bezpieczeństwa i wydajności wyrobów medycznych
- Kroki prowadzące do uzyskania oznakowania CE zgodnie z unijnym rozporządzeniem MDR dla przetworników IBP
- Rola Notyfikowanych Organów w weryfikacji zgodności z wymogami CE
- Samodzielna certyfikacja a ocena przez podmiot trzeci dla urządzeń klasy IIa
-
Standardy biokompatybilności ISO 10993 i zarządzania ryzykiem ISO 14971
- Dlaczego norma ISO 10993 jest ważna dla komponentów wchodzących w kontakt z pacjentem w systemach IBP
- Ocena ryzyka cytotoksyczności, uczulenia i podrażnień
- Studium przypadku: Błędy w doborze materiałów prowadzące do braku zgodności
- Integrowanie badań biokompatybilności z zarządzaniem ryzykiem zgodnie z normą ISO 14971
- Zezwolenie FDA i globalne wymagania regulacyjne dla przetworników IBP
- Zarządzanie jakością i certyfikaty produkcyjne dla dostawców przetworników IBP
-
Często Zadawane Pytania (FAQ)
- Do czego służy dokładnie przetwornik IBP?
- Dlaczego certyfikat CE jest ważny dla transducerów IBP?
- Jak normy ISO 10993 pomagają w produkcji transducerów IBP?
- Jaką rolę odgrywają upoważnione organy w zakresie zgodności z dyrektywą CE?
- Dlaczego norma ISO 13485 jest ważna dla zarządzania jakością w produkcji transducerów IBP?