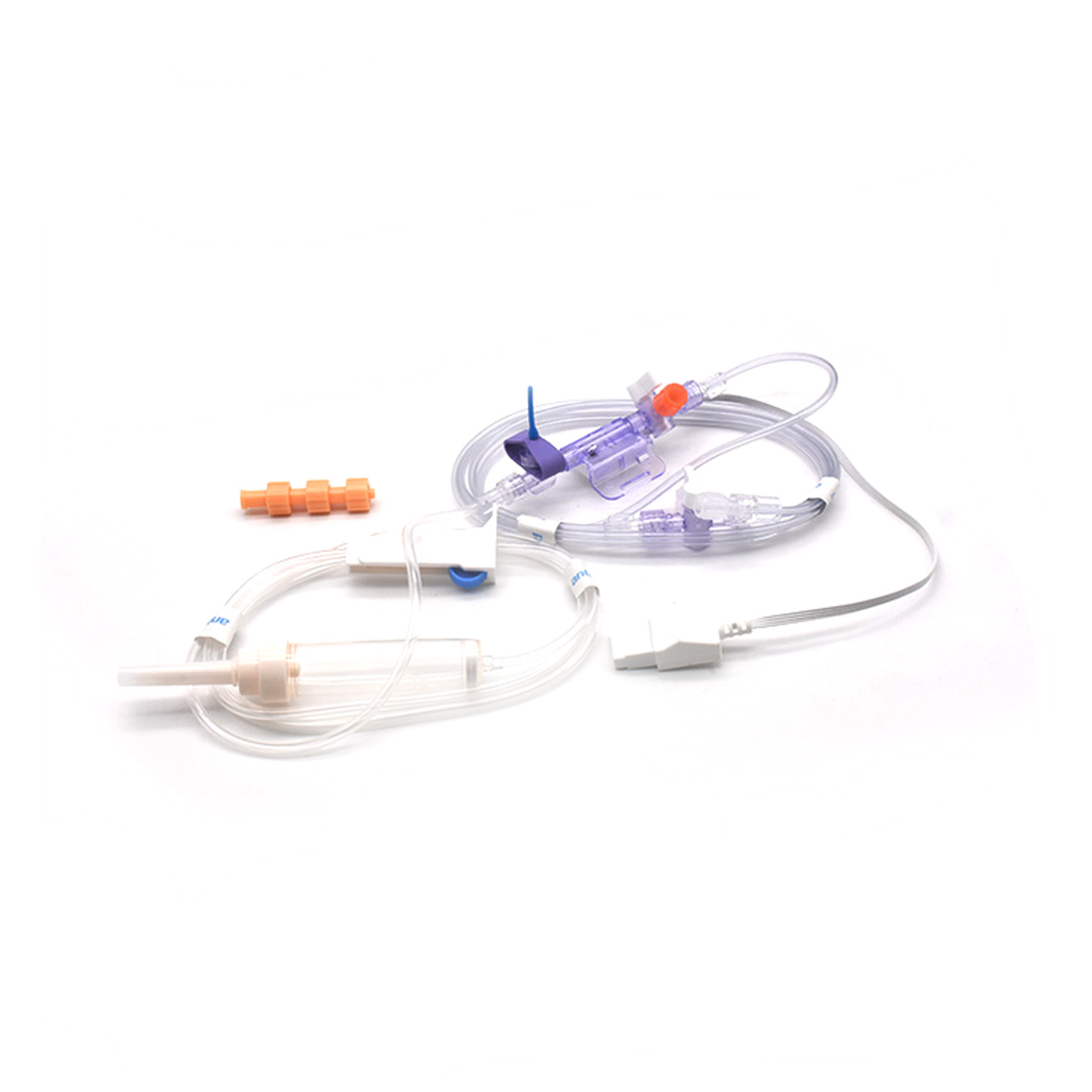
Inzicht in IBP-transducers en hun klinische betekenis
Wat is een IBP-transducator en hoe werkt deze?
Invasieve bloeddruksensoren (IBP) zijn medische apparaten die daadwerkelijke drukmetingen uit arteriën verkrijgen via katheters en deze omzetten in elektrische signalen, zodat artsen in real time kunnen zien wat er gebeurt. Deze apparaten beschikken doorgaans over steriele sensoren die zelfs de kleinste veranderingen in drukniveau kunnen detecteren, en sturen al deze informatie via vloeistofgevulde systemen naar de monitors. De nieuwere modellen halen een nauwkeurigheid van ongeveer plus of min 2 mmHg, wat van groot belang is bij het opsporen van gevaarlijke dalingen of pieken in de bloeddruk tijdens operaties. Onderzoek dat vorig jaar werd gepubliceerd in het Journal of Critical Care Medicine toonde ook iets interessants aan – patiënten die met deze nauwkeurige sensoren werden gemonitord, hadden hun lage bloeddrukepisodes 42% sneller gedetecteerd dan met reguliere niet-invasieve technieken. Dit soort vroegtijdige waarschuwing maakt een groot verschil in operatiekamers, waar elke seconde telt.
Klinisch belang van nauwkeurige invasieve bloeddrukmonitoring
Voor patiënten die vasopressoren nodig hebben of hulp bij het stabiliseren van hun circulatie, wordt invasieve bloeddrukmonitoring (IBP) nog steeds beschouwd als de beste aanpak. Oscillometrische methoden zijn onvoldoende wanneer we beat-by-beat-metingen nodig hebben, zoals in gevallen van septische shock, na ernstige ongevallen of tijdens hartoperaties. Artsen vertrouwen op de snelle reactietijd van deze monitoren, meestal binnen één seconde, om beslissingen te nemen over bijvoorbeeld vloeistoftoediening. Uit klinische ervaring blijkt dat zelfs vijf minuten wachten om bloeddrukafwijkingen te corrigeren, kan leiden tot ongeveer 15% hogere sterftecijfers. Een recente blik op icp-praktijken toonde ook iets interessants aan: ziekenhuizen die standaard IBP-transduceropstellingen invoerden, zagen ongeveer 31% minder fouten bij het aanpassen van medicatie op hun intensive care-afdelingen.
CE-certificering en EU MDR-naleving voor IBP-transducers
Wat CE-certificering betekent voor de veiligheid en prestaties van medische hulpmiddelen
CE-certificering verkrijgen volgens de EU-verordening inzake medische hulpmiddelen (MDR) betekent in principe dat een IBP-transducer strenge veiligheidscontroles heeft doorstaan die nodig zijn om in Europa te mogen verkopen. De CE-markering geeft aan dat het product voldoet aan belangrijke normen zoals IEC 60601 voor medische elektronische apparatuur en ook voldoet aan de risicobeheersregels uit ISO 14971. Voor apparaten die voldoen aan de MDR-regelgeving, is er een hele procedure om te testen hoe goed materialen die contact maken met patiënten veilig samenwerken, zodat ze geen celbeschadiging of irriterende problemen veroorzaken. De meeste bloeddrukmeterapparaten vallen onder categorie IIa of hoger, wat betekent dat fabrikanten niet zelfstandig conformiteit kunnen verklaren, maar onafhankelijke experts nodig hebben om alles te verifiëren voordat het product op de markt komt.
Stappen om CE-markering te verkrijgen onder de EU-MDR voor IBP-transducers
- Classificeer het apparaat : Bepaal de risicoclassificatie (meestal klasse IIa voor IBP-transducers) overeenkomstig bijlage VIII van de MDR.
- Implementeer een kwaliteitssysteem (QMS) : Richt een op ISO 13485-gecertificeerd kwaliteitsmanagementsysteem in.
- Stel technische documentatie samen : Compileer ontwerpspecificaties, risicobeoordelingen en rapporten van klinische evaluaties.
- Betrek een aangemelde instantie : Voor apparaten van klasse IIa en hoger, onderga audits van technische dossiers en productieprocessen.
- Plaats CE-markering : Na de conformiteitsbeoordeling, registreer het hulpmiddel in EUDAMED. Volgens SimplerQMS is 89 procent van de vertragingen te wijten aan onvolledige klinische evaluaties of verouderde QMS-documentatie.
Rol van aangemelde instanties bij het valideren van CE-conformiteit
De aangemelde instanties, of kortweg NB's, fungeren als onafhankelijke auditors bij medische hulpmiddelen die zijn ingedeeld als hoger risico. Hun taak bestaat eruit alle technische documenten te controleren en daadwerkelijk aanwezig te zijn op productielocaties voor inspecties. Bij IBP-transducers controleren deze instanties of alles voldoet aan de eisen zoals uiteengezet in MDR-bijlage II. Dit omvat het waarborgen van correcte softwarevalidatie voor digitale versies en het naleven van strikte steriliteitsnormen voor wegwerpartikelen. Uit een recente enquête uit 2023 blijkt dat de meeste problemen die door NB's worden gevonden, voortkomen uit twee hoofdgebieden: bedrijven die geen solide strategieën hebben voor post-markt surveillance, of onvoldoende biocompatibiliteitsgegevens verstrekken. Deze bevindingen onderstrepen waar fabrikanten hun aandacht op moeten richten om audits succesvol te doorlopen.
Zelfcertificering versus derdepartijbeoordeling voor klasse IIa-apparaten
Hoewel klasse I niet-steriele hulpmiddelen zichzelf kunnen certificeren, is voor klasse IIa IBP-transducers verplicht toezicht door een aangemelde instantie vereist. Zelfcertificering is alleen van toepassing op laagrisico accessoires zoals herbruikbare kabels; drukgevoelige componenten vereisen een volledige externe beoordeling. Recente MDR-updates schrijven jaarlijkse audits door de aangemelde instantie voor fabrikanten van klasse IIa voor, waardoor het aantal niet-conforme producten op de markt sinds 2021 met 34% is gedaald.
ISO 10993 Biocompatibiliteit en ISO 14971 Risicobeheernormen
Waarom ISO 10993 belangrijk is voor patiëntcontactcomponenten in IBP-systemen
Voor IBP-transductoren die daadwerkelijk contact maken met patiënten, is het volgen van de ISO 10993-normen vrijwel verplicht als we die vervelende biologische reacties willen voorkomen. Het hele doel van deze norm is om te achterhalen wat er gebeurt wanneer mensen gedurende lange periodes worden blootgesteld aan allerlei materialen, zoals kunststofonderdelen, lijm gebruikt bij assemblage en diverse sensordelen. Uit onderzoek dat vorig jaar werd gepubliceerd, blijkt dat ongeveer één op de vijf gevallen van huidirritatie die in ziekenhuizen worden gemeld, afkomstig zijn van medische hulpmiddelen gemaakt van materialen die niet voldoen aan de eisen van ISO 10993-10. Bedrijven die deze aspecten integreren in hun productieproces via een correct opgezet Biologisch Evaluatieplan lopen over het algemeen minder problemen tegen, omdat ze testen hoe verschillende materialen reageren bij contact met echte lichaamsvloeistoffen en weefsels over tijd.
Beoordeling van cytotoxiciteit, sensibilisatie en irriteringsrisico's
ISO 10993 vereist drie basisproeven voor materialen van IBP-transductoren:
- Cytotoxiciteit (ISO 10993-5): Meet de celviabiliteit na blootstelling aan materialen (’70% celdood toegestaan)
- Sensibilisatie (ISO 10993-10): Evalueert het risico op allergische reacties met behulp van maximale proeven bij kaviaaltjes
- Irritatie (ISO 10993-10): Beoordeelt het risico op lokale ontstekingen via epidermale plamuurtesten
Apparaten die een test niet halen, lopen een 89% hoger risico op terugroepactie door de FDA in vergelijking met volledig conformerende alternatieven (Medical Device Reporting, 2022).
Casestudie: Mislukte materiaalkeuze leidend tot non-conformiteit
Uit een EU MDR-audit uit 2021 bleek dat 32% van de afgewezen IBP-transductoren siliciumbuisjes gebruikten zonder ISO 10993-33-certificering voor langdurig bloedcontact. De terugroepactie van de fabrikant, goed voor $2,7 miljoen, was het gevolg van:
- De veronderstelling dat "medisch-rang" materialen automatisch voldoen aan biocompatibiliteitsnormen
- Het overslaan van versnelde verouderingstests voor risico's van chemische uitloging
- Onvoldoende documentatie van de sterilisatie-impact op materiaalveiligheid
Integratie van biocompatibiliteitstesten met risicobeheersing volgens ISO 14971
Het combineren van ISO 10993-gegevens met het risicokader van ISO 14971 stelt fabrikanten in staat om:
- Biologische risico's kwantificeren aan de hand van gevaarlijkheidsseriousheid/kansmatrices
- Controles implementeren zoals materiaalvervanging of biocompatibele coatings
- Nasporing van naverkoop bijwerkingen via processen die afgestemd zijn op ISO 13485
Deze tweeledige aanpak vermindert ontwerpwijzigingen in een laat stadium met 41% ten opzichte van alleenstaande biocompatibiliteitstesten (MedTech Quality Benchmark, 2023).
FDA-goedkeuring en wereldwijde regelgevingsvereisten voor IBP-transducers
FDA 510(k)-goedkeuringsprocedure en substantiële gelijkwaardigheid
Voor fabrikanten van medische hulpmiddelen betekent het verkrijgen van goedkeuring via het FDA 510(k)-programma aantonen dat hun product in wezen gelijkwaardig is aan een product dat al op de markt is. Volgens branchegegevens werd ongeveer driekwart van alle apparaten op deze manier goedgekeurd in 2023. Het voldoen aan deze eisen vereist uitgebreide testwerkzaamheden en strikte naleving van de regels inzake kwaliteitssystemen uit 21 CFR Part 820. Bedrijven met ISO 13485-certificering zien hun aanvragen doorgaans ongeveer 30 procent sneller door de FDA worden verwerkt. Dit lijkt te komen door beter georganiseerde administratie en documentatiepraktijken die aansluiten bij wat regelgevers verwachten tijdens de beoordeling.
Uitdagingen bij harmonisatie: verschillen tussen de EU MDR, de FDA en andere markten
Divergerende regionale eisen creëren complexiteit voor naleving:
- De EU MDR verplicht tot klinische evaluaties voor CE-markering
- De FDA benadrukt op voorganger gebaseerde gelijkwaardigheid
- De Japanse PMDA vereist naleving van ISO 80369-6 voor neuraxiale aansluitingen
- Brazil's ANVISA handhaaft lokale biocompatibiliteitstesten
Deze verschillen dwingen fabrikanten ertoe om 2 tot 3 apparaatvarianten in stand te houden, wat de ontwikkelkosten per product verhoogt met 500.000 tot 1,2 miljoen dollar (Global MedTech Compliance Report 2024).
Trend: Toenemende strengheid van eisen voor post-marktsurveillantie
Regelgevers eisen nu gegevens over prestaties uit de praktijk, waarbij de FDA sinds 2021 40% meer post-marktonderzoeken vereist. Fabrikanten moeten elektronische traceerbaarheidssystemen implementeren om bijwerkingen binnen 15 dagen te melden — 50% sneller dan de termijnen in 2020. De mislukkingspercentages voor surveillantie-audits zijn gestegen tot 22% in de hele sector, wat onderstreept hoe belangrijk proactief risicomanagement is (Medical Device Regulatory Journal 2023).
Kwaliteitsbeheer en productiecertificeringen voor leveranciers van IBP-transducers
Belang van ISO 13485 bij het ontwerp en de productie van IBP-transducers
De ISO 13485-certificering is erg belangrijk voor kwaliteitsmanagement bij leveranciers van IBP-transducers. Het helpt om controle te behouden over alle fasen, van ontwerp tot productie en verder tot na de introductie van producten op de markt. Wat dit onderscheidt van reguliere kwaliteitsnormen, is de manier waarop specifieke eisen voor medische hulpmiddelen worden aangepakt. Denk aan aspecten zoals het kunnen traceren van ontwerpen tot hun oorsprong, het correct functioneren van sterilisatie en besluitvorming op basis van daadwerkelijke risico's in plaats van gissingen. Gegevens uit 2023 over teruggeroepen medische hulpmiddelen tonen ook iets interessants aan: bedrijven met ISO 13485-certificering hadden ongeveer de helft minder problemen met het voldoen aan normen dan bedrijven zonder certificering. Als we het specifiek over IBP-transducers hebben, betekent het volgen van deze richtlijnen dat druksensoren en vloeistofpaden binnen strikte veiligheidsmarges blijven en gedurende hun hele levenscyclus betrouwbaar presteren in ziekenhuizen en klinieken.
Overeenstemming met goede fabricagepraktijk (GMP) in contractmanufacturering
Het volgen van de richtlijnen voor goede fabricagepraktijk (GMP) helpt contractfabrikanten om consistentie te behouden bij de productie van IBP-transductoren die voldoen aan zowel FDA- als EU MDR-normen. Waarom is GMP zo belangrijk? Het betreft drie hoofdgebieden: het bijhouden van correcte registraties, het zorgen dat apparatuur goed gekalibreerd blijft en het adequaat opleiden van personeel. Deze aspecten hebben een directe invloed op de nauwkeurigheid van de metingen van die invasieve bloeddrukmonitoren. Uit recente auditgegevens blijkt dat fabrieken die volledig conform GMP werken, ongeveer 98% slagingspercentage halen bij hun eerste poging in steriliteitstests. Dat is aanzienlijk beter dan de ongeveer 72% die wordt gezien bij bedrijven die deze regels slechts gedeeltelijk volgen. Wanneer bedrijven delen van het productieproces uitbesteden, zoals het maken van de transducermembraan of het monteren ervan, wordt het naleven van GMP nog essentiëler. Kleine fouten in cleanroomprocedures bij deze externe partijen kunnen de kwaliteit van het eindproduct ernstig verstoren.
Frequently Asked Questions (FAQ)
Wat doet een IBP-transducer precies?
IBP-transductoren zetten drukmetingen van slagaders om in elektrische signalen, waardoor de bloeddruk in real-time kan worden gemonitord tijdens kritische medische procedures.
Waarom is CE-certificering belangrijk voor IBP-transductoren?
CE-certificering garandeert dat IBP-transductoren voldoen aan strenge veiligheidsnormen die vereist zijn voor medische hulpmiddelen die in Europa worden verkocht, en beschermt zo de patiëntveiligheid.
Hoe helpen ISO 10993-normen bij de productie van IBP-transductoren?
ISO 10993-normen stellen fabrikanten in staat materialen te beoordelen op biocompatibiliteit, waardoor ongewenste biologische reacties door contact met de patiënt tot een minimum worden beperkt.
Welke rol spelen aangemelde instanties bij CE-conformiteit?
Aangemelde instanties fungeren als onafhankelijke auditors die controleren of IBP-transductoren voldoen aan de wettelijke eisen voordat ze op de markt komen.
Waarom is ISO 13485 belangrijk voor kwaliteitsmanagement bij de productie van IBP-transductoren?
ISO 13485 zorgt ervoor dat leveranciers van medische hulpmiddelen hoge normen handhaven op het gebied van ontwerp, productie en praktijken na de introductie op de markt, om de productkwaliteit en -veiligheid te waarborgen.
Inhoudsopgave
- Inzicht in IBP-transducers en hun klinische betekenis
-
CE-certificering en EU MDR-naleving voor IBP-transducers
- Wat CE-certificering betekent voor de veiligheid en prestaties van medische hulpmiddelen
- Stappen om CE-markering te verkrijgen onder de EU-MDR voor IBP-transducers
- Rol van aangemelde instanties bij het valideren van CE-conformiteit
- Zelfcertificering versus derdepartijbeoordeling voor klasse IIa-apparaten
- ISO 10993 Biocompatibiliteit en ISO 14971 Risicobeheernormen
- FDA-goedkeuring en wereldwijde regelgevingsvereisten voor IBP-transducers
- Kwaliteitsbeheer en productiecertificeringen voor leveranciers van IBP-transducers
-
Frequently Asked Questions (FAQ)
- Wat doet een IBP-transducer precies?
- Waarom is CE-certificering belangrijk voor IBP-transductoren?
- Hoe helpen ISO 10993-normen bij de productie van IBP-transductoren?
- Welke rol spelen aangemelde instanties bij CE-conformiteit?
- Waarom is ISO 13485 belangrijk voor kwaliteitsmanagement bij de productie van IBP-transductoren?