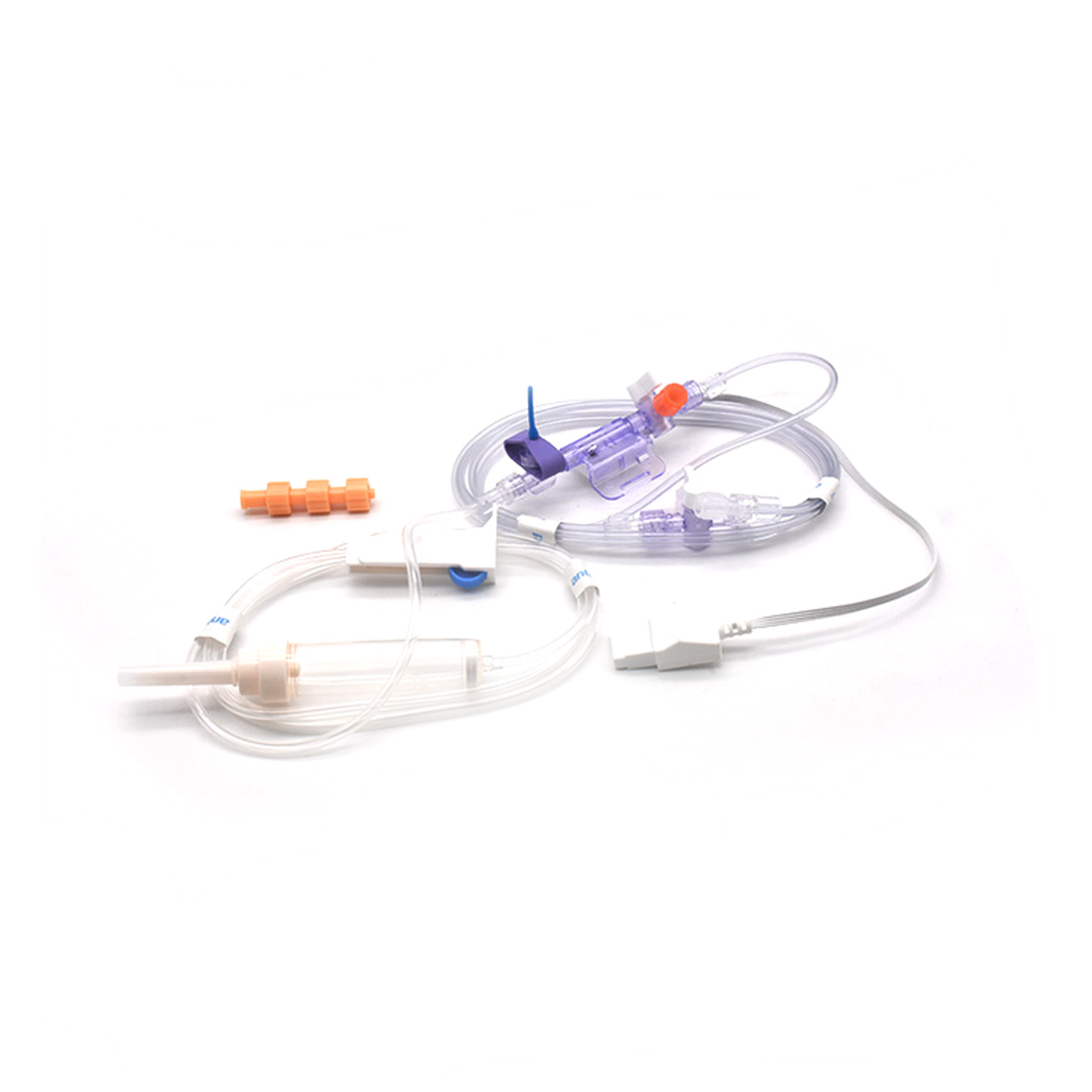
IBP transduseru izpratne un to klīniskā nozīme
Kas ir IBP pārveidotājs un kā tas darbojas?
Invasīvi asinsspiediena (IBP) pārveidotāji ir medicīniski ierīces, kas veic faktiskos spiediena mērījumus no caur katetrus savienotām artērijām un pārvērš tos par elektriskiem signāliem, lai ārsti varētu reālā laikā vērot, kas notiek. Šīm ierīcēm parasti ir sensori, kas tiek uzturēti sterilā stāvoklī, lai uztvertu pat vismazākos spiediena līmeņa svārstības, pēc tam visu šo informāciju nosūtot monitoriem, izmantojot šķidruma sistēmas. Jaunākie modeļi sasniedz aptuveni plus vai mīnus 2 mmHg precizitāti, kas ir īpaši svarīgi, lai operāciju laikā noteiktu bīstamas asinsspiediena kritumu vai skrējienu. Pērn publicēts pētījums žurnālā „Journal of Critical Care Medicine” parādīja arī kaut ko interesantu – pacientiem, kuri tika uzraudzīti ar šiem precīzajiem pārveidotājiem, zems asinsspiediens tika konstatēts 42% ātrāk salīdzinājumā ar parastām neinvazīvām metodēm. Šāda veida agrīnā brīdinājuma sistēma operāciju zālēs padara lielu atšķirību, kur sekundes ir svarīgas.
Precīzas invazīvas asinsspiediena uzraudzības klīniskā nozīme
Pacientiem, kuriem nepieciešami vazopresori vai palīdzība cirkulācijas stabilizēšanā, invazīvā asinsspiediena (IBP) monitorings joprojām tiek uzskatīts par labāko pieeju. Oscilometriskās metodes vienkārši nav pietiekamas, kad mums vajadzīgi katras sirdsdarbības dati, kas ir tik svarīgi sepsi šoka gadījumos, pēc smagiem negadījumiem vai sirds operāciju laikā. Ārsti paļaujas uz šo monitoru reakcijas ātrumu, parasti zem vienas sekundes, lai izlemtu, piemēram, par šķidruma ievadīšanu. Mēs esam redzējuši no klīniskās pieredzes, ka pat piecu minūšu gaidīšana, lai novērstu asinsspiediena problēmas, var izraisīt aptuveni par 15% augstāku nāves risku. Nesen veikts ICU prakses pārskats parādīja arī kaut ko interesantu: slimnīcas, kas ieviesa standarta IBP transduseru iestatījumus, intensīvās aprūpes nodaļās pieredzēja aptuveni par 31% mazāk kļūdu, regulējot medikamentus.
CE sertifikācija un ES MDR atbilstība IBP transduseriem
Ko nozīmē CE sertifikācija medicīnisko ierīču drošībai un veiktspējai
Iegūstot ES atbilstības sertifikāciju saskaņā ar Eiropas Savienības medicīnisko ierīču regulu (MDR), būtībā tiek apstiprināts, ka IBP transducers izturējis stingrus drošības pārbaudes testus, kas nepieciešami, lai to pārdotu Eiropā. ES atbilstības marķējums liecina, ka produkts atbilst svarīgiem standartiem, piemēram, IEC 60601 medicīniskajai elektronikas aprīkojumam, kā arī ievēro ISO 14971 noteiktos risku pārvaldības noteikumus. Ierīcēm, kas atbilst MDR noteikumiem, ir paredzēts visaptverošs procesa tests, lai novērtētu, cik droši savstarpēji darbojas materiāli, kas saskaras ar pacientu, nodrošinot, ka tie neizraisīs šūnu bojājumus vai kairinājumu. Lielākā daļa asinsspiediena monitoru ierīču ietilpst IIa klases kategorijā vai augstākā, kas nozīmē, ka ražotāji nevar vienkārši paši deklarēt atbilstību, bet tiem jāpievieno neatkarīgi eksperti, lai pirms tirgū izlaišanas verificētu, ka viss darbojas pareizi.
Soļi, lai iegūtu ES atbilstības marķējumu saskaņā ar ES MDR priekš IBP transduktoriem
- Klasificēt ierīci : Noteikt riska klasifikāciju (parasti IIa klase priekš IBP transduktoriem) saskaņā ar MDR VIII pielikumu.
- Ieviest kvalitātes vadības sistēmu (QMS) : Izveidot ISO 13485 sertificētu kvalitātes pārvaldības sistēmu.
- Sagatavot tehnisko dokumentāciju : Sastādīt dizaina specifikācijas, riska novērtējumus un klīniskās novērtēšanas atskaites.
- Iesaistīt Paziņoto struktūru : Priekš IIa+ klases ierīcēm veikt tehniskās dokumentācijas un ražošanas procesu revīzijas.
- Pielīmēt CE zīmi : Pēc atbilstības novērtējuma reģistrēt ierīci EUDAMED. Saskaņā ar SimplerQMS 89 % aizkavēšanās rodas dēļ nepilnīgām klīniskajām novērtēšanām vai novecojušai KPS dokumentācijai.
Paziņoto struktūru loma CE atbilstības validēšanā
Paziņotās struktūras, saīsināti PS, darbojas kā neatkarīgi revizori, attiecoties uz medicīniskajiem līdzekļiem, kas klasificēti kā augstāka riska. To uzdevums ietver visu tehnisko dokumentu izpēti un faktiskas vizītes ražošanas objektos, lai veiktu pārbaudes. Konkrēti aplūkojot IBP transduserus, šīs struktūras pārbauda, vai viss atbilst MDR II pielikumā izklāstītajām prasībām. Tas ietver arī pārliecināšanos, ka digitālajām versijām ir veikta pienācīga programmatūras validācija un vienreizlietojamie izstrādājumi atbilst stingrām sterilitātes normām. Saskaņā ar nesen veiktu aptauju 2023. gadā lielākā daļa PS konstatēto problēmu saistīta ar divām galvenajām jomām: uzņēmumiem nav izstrādātas efektīvas pasākumu uzraudzības stratēģijas tirgū vai tie nepiedāvā pietiekamu bioloģiskās saderības datu apjomu. Šie secinājumi norāda uz tām jomām, kurām ražotājiem būtu jāpievērš uzmanība, lai veiksmīgi izietu revīzijas.
Pašsertifikācija pret trešās puses novērtējumu IIa klases ierīcēm
Kaut gan I klases nesterilus ierīces var sertificēt pašas, IIa klases IBP pārveidotājiem ir obligāta Paziņotā struktūras uzraudzība. Pašsertifikācija attiecas tikai uz zema riska piederumiem, piemēram, atkārtoti izmantojamām vadu sistēmām; spiedienu uztverošajām sastāvdaļām nepieciešama pilna trešās puses pārbaude. Jaunākie MDR grozījumi paredz ik gadu veikt Paziņotās struktūras audits IIa klases ražotājiem, tādējādi kopš 2021. gada samazinot neatbilstošu produktu parādīšanos tirgū par 34%.
ISO 10993 bioloģiskās savietojamības un ISO 14971 risku vadības standarti
Kāpēc ISO 10993 ir svarīga pacientu kontaktkomponentēm IBP sistēmās
Attiecībā uz IBP transduseriem, kas faktiski saskaras ar pacientiem, ISO 10993 standartu ievērošana ir gandrīz obligāta, ja vēlamies izvairīties no nepatīkamām bioloģiskām reakcijām. Šī standarta mērķis ir noskaidrot, kas notiek, kad cilvēki ilgstoši tiek pakļauti dažādiem materiāliem, piemēram, plastmasas daļām, līmim, ko izmanto montāžā, un dažādiem sensora komponentiem. Saskaņā ar pērn publicētajiem pētījumiem aptuveni katrs piektais ādas kairinājuma gadījums, par kuru ziņots slimnīcās, bija saistīts ar medicīniskajiem instrumentiem, kas izgatavoti no materiāliem, kuri neatbilda ISO 10993-10 prasībām. Uzņēmumi, kas šos aspektus iekļauj savā ražošanas procesā, izmantojot pienācīgus bioloģiskās novērtēšanas plānus, parasti saskaras ar mazāk problēmām, jo tie pārbauda, kā dažādi materiāli reaģē, saskaroties ar patiesiem ķermeņa šķidrumiem un audiem laika gaitā.
Citolītiskuma, sensitizācijas un kairinājuma risku novērtēšana
ISO 10993 attiecībā uz IBP transduseru materiāliem prasa trīs pamatpārbaudes:
- Citolītiskums (ISO 10993-5): Novērtē šūnu dzīvotspēju pēc materiāla iedarbības (atļauts līdz 70% šūnu nāvei)
- Sensibilizācija (ISO 10993-10): Novērtē alerģiskas reakcijas potenciālu, izmantojot trušu maksimizācijas testus
- Irritācija (ISO 10993-10): Novērtē lokalizētas iekaisuma riska pakāpi, veicot epidēmas plāksteru testēšanu
Ierīces, kas neiziet kādu no testiem, saskaras ar par 89% augstāku ASV FDA atsaukumu biežumu salīdzinājumā ar pilnībā atbilstošiem alternatīviem risinājumiem (Medicīnisko ierīču ziņošana, 2022).
Pielietojuma piemērs: Materiālu izvēles kļūdas, kas noved pie neatbilstības
2021. gada ES MDR audits atklāja, ka 32% noraidītajām IBP transduseru ierīcēm tika izmantota silikona caurule, kurai trūka ISO 10993-33 sertifikācijas ilgstošai asins saskarei. Ražotāja 2,7 miljonu dolāru atsaukums bija saistīts ar:
- Pieņēmumu, ka „medicīniskā klases“ materiāli automātiski atbilst bioloģiskās savietojamības standartiem
- Ātrināto novecošanas testu izlaišanu attiecībā uz risku ķīmisko vielu izskalošanās dēļ
- Nepietiekama dokumentācija par sterilizācijas ietekmi uz materiālu drošumu
Bioloģiskās saderības testēšanas integrēšana ar risku pārvaldību saskaņā ar ISO 14971
ISO 10993 datu kombinēšana ar ISO 14971 riska struktūru ļauj ražotājiem:
- Kvantitatīvi novērtēt bioloģiskos riskus, izmantojot bīstamības smaguma/probabilitātes matricas
- Ieviest kontroles pasākumus, piemēram, materiālu aizvietošanu vai bioloģiski saderīgas pārklājumslāņus
- Uzraudzīt tirgus posma negatīvos notikumus, izmantojot ar ISO 13485 saskaņotas procedūras
Šis divējāds pieeja samazina dizaina izmaiņas vēlākos posmos par 41% salīdzinājumā ar atsevišķu bioloģiskās saderības testēšanu (MedTech kvalitātes orientieris, 2023).
FDA apstiprinājums un globālie regulatorie prasījumi IBP transduseriem
FDA 510(k) apstiprinājuma process un būtiskā līdzvērtība
Medicīnisko ierīču ražotājiem, lai iegūtu atļauju caur FDA 510(k) programmu, ir jāpierāda, ka to produkts ir būtiski līdzvērtīgs kaut kam, kas jau ir tirgū. Saskaņā ar nozares datiem 2023. gadā apmēram trīs ceturtdaļas visu ierīču tika apstiprinātas tieši šādā veidā. Šo prasību izpildei nepieciešama nopietna testēšanas darbība un stingra atbilstība kvalitātes sistēmas noteikumiem 21 CFR 820. paragrāfā. Uzņēmumi, kuriem ir ISO 13485 sertifikācija, parasti redz, ka FDA viņu pieteikumus apstrādā aptuveni par 30 procentiem ātrāk. Šķiet, ka tas saistīts ar labāk organizētiem dokumentiem un dokumentācijas praksi, kas atbilst tam, ko regulatori gaida redzēt pārskatīšanas laikā.
Harmonizācijas izaicinājumi: atšķirības starp ES MDR, FDA un citiem tirgiem
Atšķirīgas reģionālās prasības rada atbilstības sarežģītību:
- ES MDR paredz klīniskās novērtēšanas CE marķējumam
- FDA uzsvērt predikāta bāzētu līdzvērtību
- Japānas PMDA prasa atbilstību ISO 80369-6 neiraksijām savienojumiem
- Brazīlijas ANVISA uzliek obligātu vietējo bioloģiskās saderības testēšanu
Šie atšķirības piespiež ražotājus uzturēt 2–3 ierīču variantus, palielinot izstrādes izmaksas par 500 tūkstošiem – 1,2 miljoniem USD par produktu (Global MedTech Compliance Report 2024).
Tendence: ASV pēcpārdošanas uzraudzības prasību pastiprināšanās
Regulatori tagad prasa reālās lietošanas rezultātu datus, pieprasot FDA pēcpārdošanas pētījumus par 40% vairāk nekā kopš 2021. gada. Ražotājiem ir jāievieš elektroniskas izsekojamības sistēmas, lai ziņotu par negatīviem notikumiem 15 dienu laikā — par 50% ātrāk nekā 2020. gada termiņi. Uzraudzības auditu atteikumu likme ir pieaugusi līdz 22% visā nozarē, kas uzsvērti norāda uz nepieciešamību pēc proaktīvas riska pārvaldības (Medical Device Regulatory Journal 2023).
Kvalitātes pārvaldība un ražošanas sertifikācijas IBP transduseru piegādātājiem
ISO 13485 nozīme IBP transduseru dizainā un ražošanā
ISO 13485 sertifikācija ir ļoti svarīga kvalitātes pārvaldībai starp IBP transduseru piegādātājiem. Tā palīdz uzturēt kontroli visās posmos — sākot no dizaina, ražošanas un līdz pat tam, kas notiek pēc tam, kad produkti nonāk tirgū. To, kas atšķir šo standartu no parastajiem kvalitātes standartiem, ir tā specifiskās prasības medicīniskajiem ierīcēm. Iedomājieties lietas, piemēram, spēju sekot dizainam līdz tā izcelsmes avotam, nodrošināt, ka sterilizācija darbojas pareizi, un lēmumu pieņemšanu, balstoties uz faktiskiem riskiem, nevis minējumiem. Skatoties uz 2023. gada datiem par atsauktajām medicīniskajām ierīcēm, redzams arī kaut kas interesants. Uzņēmumiem ar ISO 13485 sertifikāciju bija aptuveni pusi mazāk problēmu ar standartu ievērošanu salīdzinājumā ar tiem, kam tāda nav. Kad mēs konkrēti runājam par IBP transduseriem, šo norādījumu ievērošana nozīmē, ka spiediena sensori un šķidruma plūsmas maršruti paliek stingros drošības robežos un uzticami darbojas visā to dzīves ciklā slimnīcās un klīnikās.
Labas ražošanas prakses (GMP) atbilstība uzticētajā ražošanā
Ievērojot Labas ražošanas prakses (GMP) norādījumus, līgumražotāji var nodrošināt vienmērību IBP transduseru ražošanā, kas atbilst gan ASV FDA, gan ES MDR standartiem. Kāpēc GMP ir tik svarīga? Tā aptver trīs galvenas jomas: pienācīgu dokumentāciju, aprīkojuma kalibrācijas uzturēšanu un personāla apmācību. Šie aspekti tieši ietekmē invazīvo asinsspiediena monitoru mērījumu precizitāti. Analizējot neseno revīziju datus, konstatēts, ka iekārtām, kas pilnībā atbilst GMP prasībām, sterilumtestos pirmajā reizē izdodas panākt aptuveni 98 % panākumu likmi. Tas ir ievērojami augstāk nekā aptuveni 72 %, ko novēro vietās, kas šīs prasības ievēro tikai daļēji. Kad uzņēmumi izsaista trešās puses ražošanas posmus, piemēram, transdusera diafragmu izgatavošanu vai montāžu, GMP ievērošana kļūst vēl būtiskāka. Nelielas kļūdas tīrkameru procedūrā šādās trešās puses vietās var nopietni ietekmēt gala produkta kvalitāti.
Dažkārt uzdots jautājumi
Ko īsti dara IBP transdusers?
IBP transduseri pārveido spiediena rādījumus no artērijām elektriskos signālos, ļaujot reāllaikā monitorēt asinsspiediun kritisku medicīnisko procedūru laikā.
Kāpēc CE sertifikācija ir svarīga IBP transduceriem?
CE sertifikācija nodrošina, ka IBP transduseri atbilst stingriem drošības standartiem, kas nepieciešami medicīniskajiem ierīcēm, kas tiek pārdotas Eiropā, aizsargājot pacientu drošību.
Kā ISO 10993 standarti palīdz ražojot IBP transducerus?
ISO 10993 standarti ļauj ražotājiem novērtēt materiālu bioloģisko savietojamību, minimizējot negatīvas bioloģiskas reakcijas, kas rodas saskarē ar pacientu.
Kāda loma Paziņotajām struktūrām ir CE atbilstībā?
Paziņotās struktūras darbojas kā neatkarīgi revizori, apstiprinot, ka IBP transduseri atbilst regulatīvajām prasībām pirms to izlaišanas tirgū.
Kāpēc ISO 13485 ir svarīga kvalitātes vadībai IBP transduceru ražošanā?
ISO 13485 nodrošina, ka medicīnisko ierīču piegādātāji uztur augstus standartus visā projektēšanas, ražošanas un pēcpārdošanas procesā, lai garantētu produkta kvalitāti un drošību.
Satura rādītājs
- IBP transduseru izpratne un to klīniskā nozīme
- CE sertifikācija un ES MDR atbilstība IBP transduseriem
- ISO 10993 bioloģiskās savietojamības un ISO 14971 risku vadības standarti
- FDA apstiprinājums un globālie regulatorie prasījumi IBP transduseriem
- Kvalitātes pārvaldība un ražošanas sertifikācijas IBP transduseru piegādātājiem
- Dažkārt uzdots jautājumi