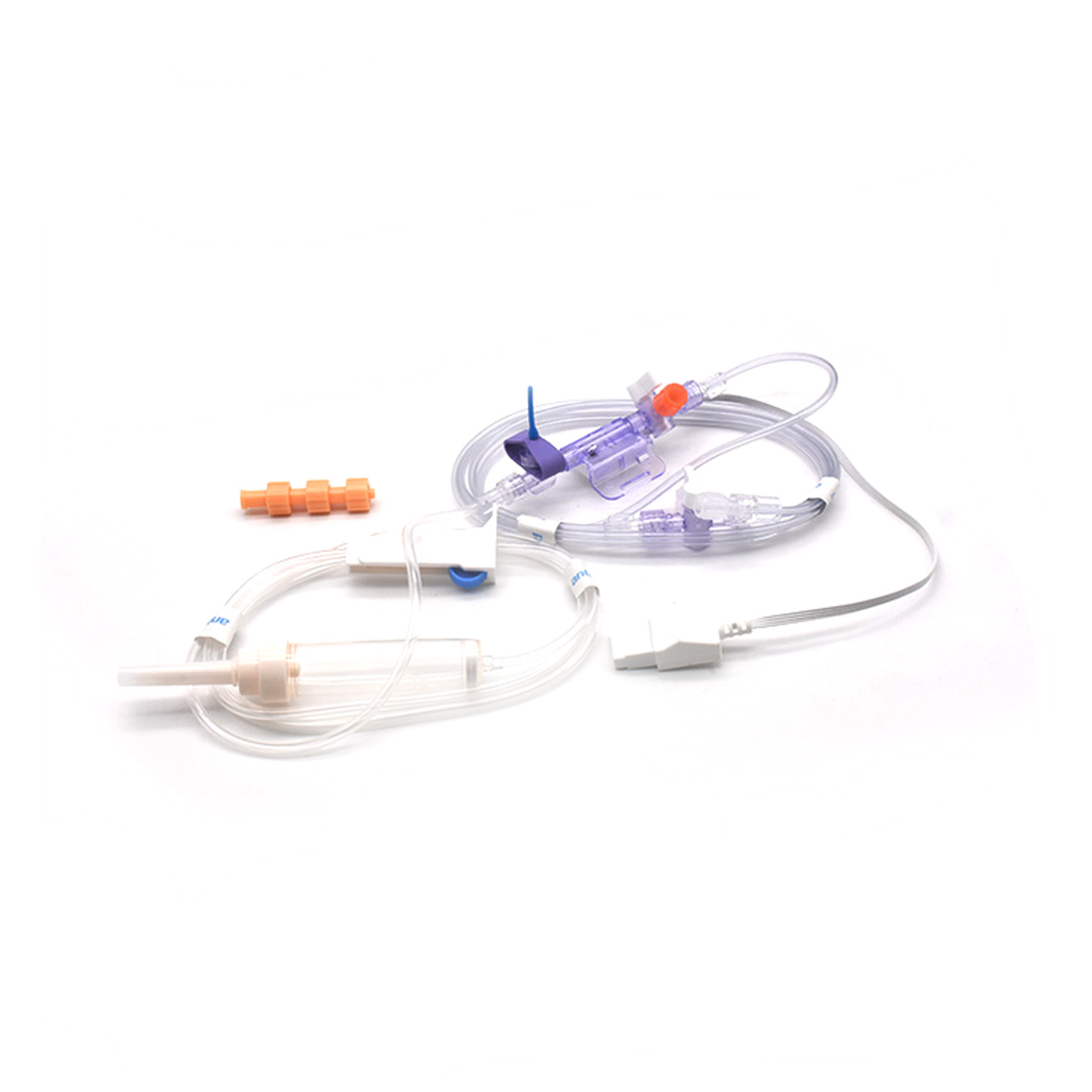
Memahami Transduser IBP dan Signifikansi Klinisnya
Apa itu transduser IBP dan bagaimana cara kerjanya?
Transduser tekanan darah invasif (IBP) adalah perangkat medis yang mengambil pembacaan tekanan aktual dari arteri yang terhubung melalui kateter dan mengubahnya menjadi sinyal listrik agar dokter dapat memantau kondisi secara waktu nyata. Perangkat ini umumnya memiliki sensor yang dijaga steril untuk mendeteksi bahkan perubahan tekanan paling kecil, lalu mengirimkan semua informasi tersebut ke monitor melalui sistem yang diisi cairan. Model-model terbaru mencapai akurasi sekitar plus minus 2 mmHg, yang sangat penting untuk mendeteksi penurunan atau lonjakan tekanan darah berbahaya selama operasi. Penelitian yang diterbitkan tahun lalu dalam Journal of Critical Care Medicine menunjukkan temuan menarik juga—pasien yang dipantau dengan transduser presisi ini mengalami deteksi episode tekanan darah rendah 42% lebih cepat dibandingkan teknik non-invasif biasa. Peringatan dini semacam ini memberikan dampak besar di ruang operasi, di mana setiap detik sangat berarti.
Pentingnya Klinis Pemantauan Tekanan Darah Invasif yang Akurat
Bagi pasien yang memerlukan vasopresor atau bantuan dalam menstabilkan sirkulasi, pemantauan tekanan darah invasif (IBP) masih dianggap sebagai pendekatan terbaik. Metode osilometrik tidak mencukupi ketika kita membutuhkan pembacaan detak demi detak yang sangat penting selama kasus syok septik, setelah kecelakaan besar, atau selama operasi jantung. Dokter mengandalkan kecepatan respons monitor ini, biasanya kurang dari satu detik, untuk mengambil keputusan seperti pemberian cairan. Dari pengalaman klinis, kita telah melihat bahwa bahkan menunggu lima menit penuh untuk mengoreksi masalah tekanan darah dapat menyebabkan peningkatan angka kematian sekitar 15%. Sebuah tinjauan terhadap praktik di ICU baru-baru ini juga menunjukkan hal menarik: rumah sakit yang menerapkan setup transduser IBP standar mengalami kesalahan sekitar 31% lebih sedikit saat menyesuaikan obat di unit perawatan intensif mereka.
Sertifikasi CE dan Kepatuhan terhadap EU MDR untuk Transduser IBP
Apa Arti Sertifikasi CE bagi Keamanan dan Kinerja Alat Kesehatan
Mendapatkan sertifikasi CE sesuai dengan Peraturan Perangkat Kesehatan Uni Eropa (MDR) pada dasarnya berarti transduser IBP lolos pemeriksaan keamanan ketat yang diperlukan untuk dijual di Eropa. Penandaan CE menunjukkan bahwa produk tersebut mematuhi standar penting seperti IEC 60601 untuk peralatan elektronik medis serta memenuhi aturan manajemen risiko dari ISO 14971. Untuk perangkat yang mematuhi regulasi MDR, terdapat proses pengujian menyeluruh terhadap kinerja material yang bersentuhan langsung dengan pasien agar aman secara bersamaan, memastikan tidak menyebabkan kerusakan sel atau iritasi. Sebagian besar perangkat pemantau tekanan darah termasuk dalam kategori Kelas IIa atau di atasnya, yang berarti produsen tidak dapat hanya menyatakan kepatuhan sendiri tetapi harus melibatkan ahli independen untuk memverifikasi bahwa semua fungsi bekerja dengan benar sebelum masuk ke pasar.
Langkah-Langkah untuk Mendapatkan Penandaan CE di Bawah MDR Uni Eropa untuk Transduser IBP
- Klasifikasikan perangkat : Tentukan klasifikasi risiko (umumnya Kelas IIa untuk transduser IBP) sesuai Lampiran VIII MDR.
- Terapkan Sistem Manajemen Mutu (QMS) : Membentuk sistem manajemen mutu yang bersertifikasi ISO 13485.
- Siapkan dokumentasi teknis : Kompilasi spesifikasi desain, penilaian risiko, dan laporan evaluasi klinis.
- Libatkan Badan yang Ditunjuk : Untuk perangkat Kelas IIa+, lakukan audit terhadap dokumen teknis dan proses manufaktur.
- Tempel tanda CE : Setelah penilaian kesesuaian, daftarkan perangkat ke EUDAMED. Delapan puluh sembilan persen keterlambatan berasal dari evaluasi klinis yang tidak lengkap atau dokumentasi SMM yang sudah usang, menurut SimplerQMS.
Peran Badan yang Ditunjuk dalam Memvalidasi Kepatuhan CE
Lembaga yang Diberitahukan, atau NB untuk singkatnya, berperan sebagai auditor independen dalam hal perangkat medis yang diklasifikasikan sebagai risiko tinggi. Tugas mereka meliputi pemeriksaan seluruh dokumen teknis dan secara langsung mengunjungi lokasi manufaktur untuk inspeksi. Ketika khusus meninjau transduser IBP, lembaga-lembaga ini memeriksa apakah semua aspek sesuai dengan persyaratan yang tercantum dalam Lampiran II MDR. Ini termasuk memastikan versi digital telah memiliki validasi perangkat lunak yang memadai dan bahwa produk sekali pakai memenuhi standar sterilitas yang ketat. Menurut survei terbaru dari tahun 2023, sebagian besar masalah yang ditemukan oleh NB berasal dari dua area utama: perusahaan yang tidak memiliki strategi pengawasan pasca-pemasaran yang kuat, atau gagal menyediakan data biokompatibilitas yang cukup. Temuan ini menyoroti aspek-aspek yang perlu menjadi fokus produsen jika ingin lulus audit dengan sukses.
Sertifikasi Mandiri vs. Penilaian Pihak Ketiga untuk Perangkat Kelas IIa
Meskipun perangkat Kelas I non-steril dapat melakukan sertifikasi mandiri, transduser IBP Kelas IIa memerlukan pengawasan wajib dari Badan yang Ditunjuk. Sertifikasi mandiri hanya berlaku untuk aksesori berisiko rendah seperti kabel yang dapat digunakan kembali; komponen sensor tekanan memerlukan tinjauan pihak ketiga secara lengkap. Pembaruan MDR terbaru mewajibkan audit tahunan oleh Badan yang Ditunjuk bagi produsen Kelas IIa, sehingga mengurangi masuknya produk yang tidak sesuai ke pasar sebesar 34% sejak tahun 2021.
Standar Biokompatibilitas ISO 10993 dan Standar Manajemen Risiko ISO 14971
Mengapa ISO 10993 Penting untuk Komponen yang Berkontak dengan Pasien dalam Sistem IBP
Untuk transduser IBP yang benar-benar bersentuhan dengan pasien, mengikuti standar ISO 10993 hampir merupakan kewajiban jika kita ingin menghindari reaksi biologis yang merugikan. Inti dari standar ini adalah untuk mengetahui apa yang terjadi ketika manusia terpapar dalam jangka waktu lama terhadap berbagai macam bahan seperti komponen plastik, lem yang digunakan dalam perakitan, dan berbagai bagian sensor. Menurut penelitian yang dipublikasikan tahun lalu, sekitar satu dari lima kasus iritasi kulit yang dilaporkan di rumah sakit berasal dari perangkat medis yang dibuat dengan bahan yang tidak memenuhi persyaratan ISO 10993-10. Perusahaan yang mengintegrasikan hal-hal ini ke dalam proses manufaktur mereka melalui Rencana Evaluasi Biologis yang tepat cenderung mengalami lebih sedikit masalah karena mereka menguji bagaimana berbagai bahan bereaksi saat bersentuhan dengan cairan tubuh dan jaringan asli dalam jangka waktu tertentu.
Evaluasi Risiko Sitotoksisitas, Sensitisasi, dan Iritasi
ISO 10993 mengharuskan tiga uji inti untuk bahan transduser IBP:
- Sitotoksisitas (ISO 10993-5): Mengukur viabilitas sel setelah terpapar bahan (’70% kematian sel diperbolehkan)
- Sensitisasi (ISO 10993-10): Mengevaluasi potensi respons alergi menggunakan uji maksimalisasi pada kelinci percobaan
- Iritasi (ISO 10993-10): Menilai risiko peradangan lokal melalui pengujian tempel epidermis
Perangkat yang gagal dalam salah satu uji menghadapi tingkat penarikan oleh FDA yang 89% lebih tinggi dibandingkan alternatif yang sepenuhnya patuh (Pelaporan Perangkat Kedokteran, 2022).
Studi Kasus: Kegagalan Pemilihan Bahan yang Mengakibatkan Ketidakpatuhan
Audit EU MDR 2021 mengungkapkan bahwa 32% transduser IBP yang ditolak menggunakan selang silikon yang tidak memiliki sertifikasi ISO 10993-33 untuk kontak darah jangka panjang. Penarikan produk senilai $2,7 juta dolar AS yang dilakukan produsen disebabkan oleh:
- Mengasumsikan bahan "kelas medis" secara otomatis memenuhi standar biokompatibilitas
- Melewatkan uji penuaan dipercepat untuk menilai risiko pelindian bahan kimia
- Dokumentasi yang tidak memadai mengenai dampak sterilisasi terhadap keamanan material
Mengintegrasikan Pengujian Kesesuaian Hayati dengan Manajemen Risiko sesuai ISO 14971
Menggabungkan data ISO 10993 dengan kerangka risiko ISO 14971 memungkinkan produsen untuk:
- Mengukur risiko biologis menggunakan matriks tingkat keparahan/probabilitas bahaya
- Menerapkan kontrol seperti penggantian material atau lapisan yang kompatibel secara hayati
- Memantau kejadian buruk pasca-pemasaran melalui proses yang selaras dengan ISO 13485
Pendekatan ganda ini mengurangi perubahan desain tahap akhir sebesar 41% dibandingkan dengan pengujian kesesuaian hayati mandiri (MedTech Quality Benchmark, 2023).
Persetujuan FDA dan Persyaratan Regulasi Global untuk Transduser IBP
Proses Persetujuan FDA 510(k) dan Kesetaraan Substansial
Bagi produsen alat kesehatan, mendapatkan persetujuan melalui program FDA 510(k) berarti menunjukkan bahwa produk mereka secara substansial setara dengan produk yang sudah dipasarkan. Sekitar tiga perempat dari seluruh alat kesehatan disetujui dengan cara ini pada tahun 2023 menurut data industri. Memenuhi persyaratan ini membutuhkan pekerjaan pengujian yang serius dan kepatuhan ketat terhadap aturan sistem mutu dalam 21 CFR Bagian 820. Perusahaan yang memiliki sertifikasi ISO 13485 cenderung mengalami proses aplikasi yang sekitar 30 persen lebih cepat di FDA. Hal ini tampaknya disebabkan oleh pengelolaan dokumen dan administrasi yang lebih terorganisir sesuai dengan harapan regulator selama proses tinjauan.
Tantangan Harmonisasi: Perbedaan antara EU MDR, FDA, dan Pasar Lainnya
Persyaratan regional yang berbeda menciptakan kompleksitas kepatuhan:
- EU MDR mewajibkan evaluasi klinis untuk penandaan CE
- FDA menekankan kesetaraan berbasis predikat
- PMDA Jepang mewajibkan kepatuhan terhadap ISO 80369-6 untuk koneksi neuraksial
- ANVISA Brasil memberlakukan pengujian biokompatibilitas lokal
Perbedaan-perbedaan ini memaksa produsen untuk mempertahankan 2–3 varian perangkat, yang meningkatkan biaya pengembangan sebesar $500.000–$1,2 juta per produk (Laporan Kepatuhan MedTech Global 2024).
Tren: Meningkatnya Ketatnya Persyaratan Pengawasan Pasca-Pemasaran
Regulator kini menuntut data kinerja dari dunia nyata, dengan persyaratan studi pasca-pemasaran FDA yang meningkat 40% sejak 2021. Produsen harus menerapkan sistem pelacakan elektronik untuk melaporkan kejadian merugikan dalam waktu 15 hari—50% lebih cepat dari tenggat waktu tahun 2020. Tingkat kegagalan audit pengawasan telah meningkat hingga 22% secara industri, menegaskan pentingnya manajemen risiko proaktif (Medical Device Regulatory Journal 2023).
Manajemen Mutu dan Sertifikasi Produksi untuk Pemasok Transduser IBP
Pentingnya ISO 13485 dalam Desain dan Produksi Transduser IBP
Sertifikasi ISO 13485 sangat penting untuk manajemen mutu di antara pemasok transduser IBP. Sertifikasi ini membantu menjaga kendali pada semua tahap, mulai dari desain hingga produksi, serta setelah produk berada di pasaran. Yang membedakan sertifikasi ini dari standar mutu biasa adalah pendekatannya terhadap kebutuhan khusus perangkat medis. Pertimbangkan hal-hal seperti kemampuan melacak desain kembali ke asalnya, memastikan proses sterilisasi berjalan dengan benar, serta pengambilan keputusan yang didasarkan pada risiko nyata, bukan tebakan. Melihat data tahun 2023 mengenai perangkat medis yang ditarik kembali menunjukkan temuan menarik juga. Perusahaan dengan sertifikasi ISO 13485 mengalami sekitar separuh lebih sedikit masalah dalam memenuhi standar dibandingkan perusahaan tanpa sertifikasi tersebut. Ketika kita berbicara secara khusus mengenai transduser IBP, mengikuti pedoman ini berarti sensor tekanan dan jalur cairan tetap berada dalam batas keselamatan yang ketat serta menunjukkan kinerja andal sepanjang siklus hidupnya di rumah sakit dan klinik.
Kesesuaian Praktik Manufaktur yang Baik (GMP) dalam Manufaktur Kontrak
Mengikuti pedoman Praktik Manufaktur yang Baik (Good Manufacturing Practice/GMP) membantu produsen kontrak menjaga konsistensi dalam memproduksi transduser IBP yang memenuhi standar FDA dan EU MDR. Mengapa GMP begitu penting? Karena GMP mencakup tiga aspek utama: menyimpan catatan yang tepat, memastikan peralatan tetap terkalibrasi, serta memberikan pelatihan yang memadai kepada staf. Aspek-aspek ini secara langsung memengaruhi ketepatan hasil pengukuran pada monitor tekanan darah invasif tersebut. Berdasarkan data audit terkini, fasilitas yang sepenuhnya patuh terhadap GMP mencapai tingkat keberhasilan sekitar 98% dalam uji sterilitas pada percobaan pertama. Angka ini jauh lebih baik dibandingkan dengan sekitar 72% yang dicatat oleh fasilitas yang hanya sebagian mengikuti aturan ini. Ketika perusahaan melakukan outsourcing terhadap bagian proses manufaktur seperti pembuatan diafragma transduser atau perakitannya, kepatuhan terhadap GMP menjadi semakin penting. Kesalahan kecil dalam prosedur ruang bersih di lokasi pihak ketiga dapat sangat merusak kualitas produk akhir.
Pertanyaan yang Sering Diajukan (FAQ)
Apa sebenarnya fungsi dari transduser IBP?
Transduser IBP mengubah pembacaan tekanan dari arteri menjadi sinyal listrik, memungkinkan pemantauan tekanan darah secara waktu nyata selama prosedur medis kritis.
Mengapa sertifikasi CE penting untuk transduser IBP?
Sertifikasi CE memastikan bahwa transduser IBP memenuhi standar keselamatan ketat yang diperlukan untuk perangkat medis yang dijual di Eropa, melindungi keselamatan pasien.
Bagaimana standar ISO 10993 membantu dalam pembuatan transduser IBP?
Standar ISO 10993 memungkinkan produsen untuk mengevaluasi bahan berdasarkan biokompatibilitasnya, meminimalkan reaksi biologis yang merugikan akibat kontak dengan pasien.
Apa peran Badan Penunjuk dalam kepatuhan CE?
Badan Penunjuk bertindak sebagai auditor independen yang memverifikasi bahwa transduser IBP memenuhi persyaratan regulasi sebelum masuk ke pasar.
Mengapa ISO 13485 penting untuk manajemen mutu dalam produksi transduser IBP?
ISO 13485 memastikan bahwa pemasok perangkat medis menjaga standar tinggi dalam desain, produksi, dan praktik pasca-pasar untuk menjamin kualitas dan keselamatan produk.
Daftar Isi
- Memahami Transduser IBP dan Signifikansi Klinisnya
- Sertifikasi CE dan Kepatuhan terhadap EU MDR untuk Transduser IBP
-
Standar Biokompatibilitas ISO 10993 dan Standar Manajemen Risiko ISO 14971
- Mengapa ISO 10993 Penting untuk Komponen yang Berkontak dengan Pasien dalam Sistem IBP
- Evaluasi Risiko Sitotoksisitas, Sensitisasi, dan Iritasi
- Studi Kasus: Kegagalan Pemilihan Bahan yang Mengakibatkan Ketidakpatuhan
- Mengintegrasikan Pengujian Kesesuaian Hayati dengan Manajemen Risiko sesuai ISO 14971
- Persetujuan FDA dan Persyaratan Regulasi Global untuk Transduser IBP
- Manajemen Mutu dan Sertifikasi Produksi untuk Pemasok Transduser IBP
- Pertanyaan yang Sering Diajukan (FAQ)