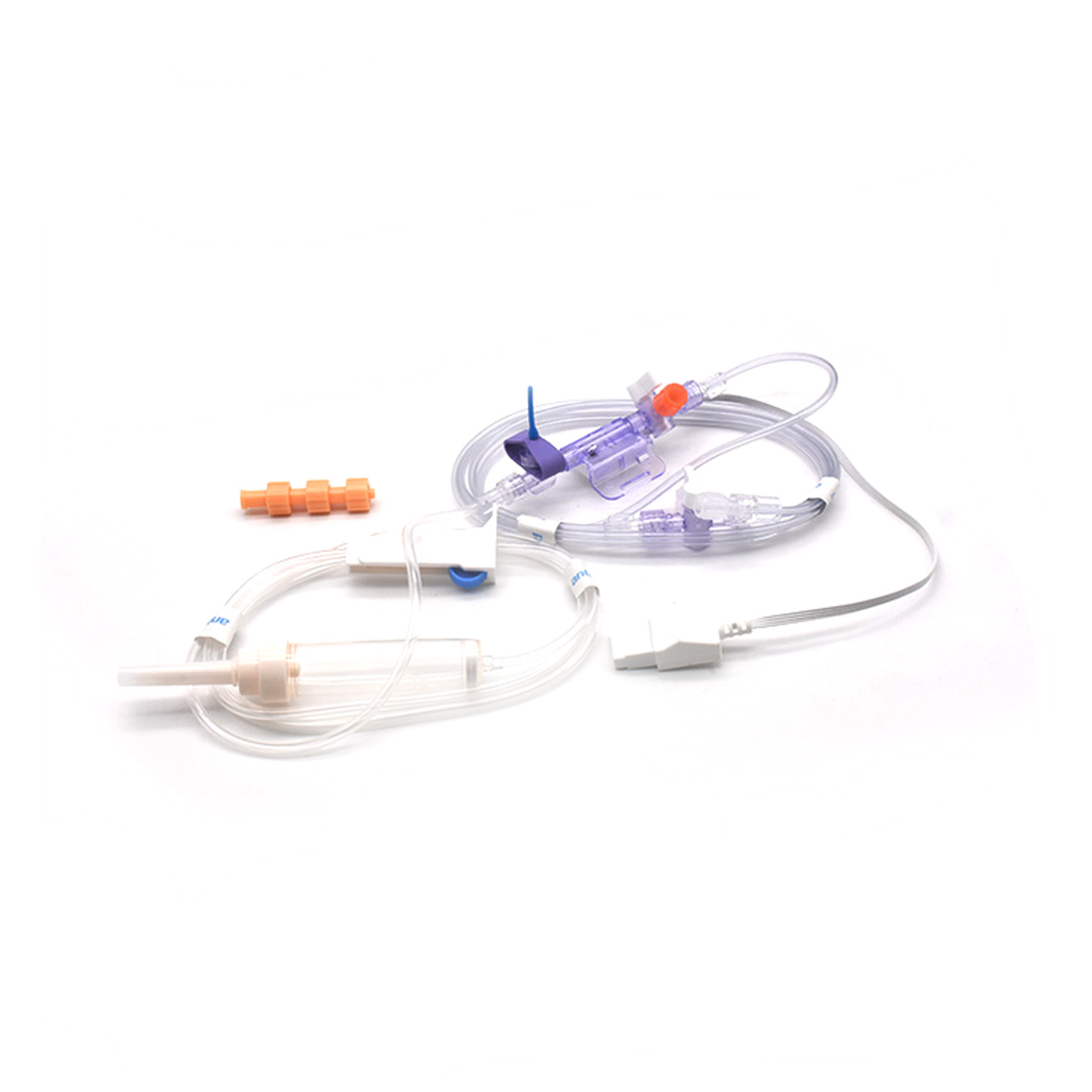
Comprendre les transducteurs IBP et leur importance clinique
Qu'est-ce qu'un transducteur IBP et comment fonctionne-t-il?
Les transducteurs de pression artérielle invasive (PAI) sont des dispositifs médicaux qui effectuent des mesures réelles de la pression dans les artères via des cathéters et convertissent ces données en signaux électriques, permettant aux médecins de surveiller en temps réel l'état du patient. Ces dispositifs intègrent généralement des capteurs stériles conçus pour détecter les plus légers changements de pression, puis transmettent ces informations aux moniteurs à l'aide de systèmes remplis de liquide. Les modèles les plus récents offrent une précision d'environ plus ou moins 2 mmHg, ce qui est crucial pour détecter rapidement les chutes ou pics dangereux de la pression artérielle pendant une intervention chirurgicale. Une étude publiée l'année dernière dans le Journal of Critical Care Medicine a révélé un résultat intéressant : les épisodes d'hypotension chez les patients surveillés avec ces transducteurs précis ont été détectés 42 % plus rapidement qu'avec les méthodes non invasives classiques. Ce type d'alerte précoce fait une grande différence en salle d'opération, où chaque seconde compte.
Importance clinique d'une surveillance invasive de la pression artérielle précise
Pour les patients ayant besoin de vasopresseurs ou d'une aide pour stabiliser leur circulation, la surveillance invasive de la pression artérielle (IBP) est toujours considérée comme la meilleure approche. Les méthodes oscillométriques ne suffisent pas lorsque nous avons besoin de mesures battant par battant, cruciales dans les cas de choc septique, après un grave accident ou pendant des interventions cardiaques. Les médecins s'appuient sur la rapidité de réaction de ces moniteurs, généralement inférieure à une seconde, pour prendre des décisions concernant notamment l'administration de fluides. L'expérience clinique a montré que même attendre cinq minutes entières avant de corriger des anomalies de la pression artérielle peut entraîner environ 15 % de décès supplémentaires. Une étude récente sur les pratiques en unité de soins intensifs a également révélé un résultat intéressant : les hôpitaux ayant mis en œuvre des configurations standardisées de transducteurs IBP ont constaté environ 31 % d'erreurs en moins lors de l'ajustement des médicaments dans leurs services de soins intensifs.
Certification CE et conformité au Règlement européen sur les dispositifs médicaux (MDR) pour les transducteurs IBP
Ce que signifie la certification CE en matière de sécurité et de performance des dispositifs médicaux
Obtenir la certification CE conformément au règlement de l'UE sur les dispositifs médicaux (MDR) signifie essentiellement qu'un transducteur IBP a passé des contrôles rigoureux en matière de sécurité nécessaires pour être vendu en Europe. Le marquage CE indique que le produit respecte des normes importantes telles que l'IEC 60601 pour les équipements électroniques médicaux et qu'il satisfait également aux règles de gestion des risques définies dans l'ISO 14971. Pour les dispositifs conformes au MDR, un processus complet est requis afin d'évaluer la compatibilité sûre des matériaux en contact avec les patients, en veillant à ce qu'ils n'entraînent ni dommages cellulaires ni irritations. La plupart des dispositifs de surveillance de la pression artérielle sont classés dans la catégorie IIa ou supérieure, ce qui signifie que les fabricants ne peuvent pas simplement déclarer la conformité eux-mêmes, mais doivent faire vérifier l'ensemble du dispositif par des experts indépendants avant sa mise sur le marché.
Étapes pour obtenir le marquage CE selon le MDR de l'UE pour les transducteurs IBP
- Classer le dispositif : Déterminer la classification du risque (généralement classe IIa pour les transducteurs IBP) conformément à l'annexe VIII du MDR.
- Mettre en œuvre un système de management de la qualité (QMS) : Mettre en place un système de management de la qualité certifié ISO 13485.
- Préparer la documentation technique : Compiler les spécifications de conception, les évaluations des risques et les rapports d'évaluation clinique.
- Saisir un organisme notifié : Pour les dispositifs de classe IIa et supérieure, subir des audits des dossiers techniques et des processus de fabrication.
- Apposer le marquage CE : Après l'évaluation de conformité, enregistrer le dispositif dans EUDAMED. Selon SimplerQMS, quatre-vingt-neuf pour cent des retards proviennent d'évaluations cliniques incomplètes ou de documentation du SMS obsolète.
Rôle des organismes notifiés dans la validation de la conformité CE
Les organismes notifiés, ou ON en abrégé, agissent comme des auditeurs indépendants pour les dispositifs médicaux classés à risque élevé. Leur mission consiste à examiner tous les documents techniques et à se rendre effectivement sur les sites de fabrication pour effectuer des inspections. En ce qui concerne spécifiquement les transducteurs IBP, ces organismes vérifient que tout est conforme aux exigences décrites à l'annexe II du règlement MDR. Cela inclut la validation adéquate des logiciels pour les versions numériques et le respect de normes strictes d'asepsie pour les produits jetables. Selon une enquête récente de 2023, la majorité des problèmes détectés par les ON proviennent de deux domaines principaux : l'absence de stratégies solides de surveillance post-commercialisation chez les entreprises, ou un défaut de fournir suffisamment de données sur la biocompatibilité. Ces constatations mettent en lumière les points sur lesquels les fabricants doivent concentrer leurs efforts afin de réussir les audits.
Auto-certification contre évaluation par un tiers pour les dispositifs de classe IIa
Bien que les dispositifs de classe I non stériles puissent être auto-certifiés, les transducteurs IBP de classe IIa nécessitent une surveillance obligatoire par un organisme notifié. L'auto-certification ne s'applique qu'aux accessoires à faible risque, tels que les câbles réutilisables ; les composants de détection de pression exigent un examen complet par un tiers. Les récentes mises à jour du RDM imposent des audits annuels par l'organisme notifié pour les fabricants de classe IIa, réduisant ainsi les introductions sur le marché non conformes de 34 % depuis 2021.
Normes de biocompatibilité ISO 10993 et norme de gestion des risques ISO 14971
Pourquoi la norme ISO 10993 est-elle importante pour les composants en contact avec le patient dans les systèmes IBP
Pour les transducteurs de PIA qui entrent effectivement en contact avec les patients, le respect des normes ISO 10993 est pratiquement obligatoire si l'on souhaite éviter les réactions biologiques indésirables. L'objectif principal de cette norme est d'évaluer ce qui se produit lorsque les personnes sont exposées pendant de longues périodes à toutes sortes de matériaux, comme des pièces en plastique, les colles utilisées lors de l'assemblage ou divers composants de capteurs. Selon des recherches publiées l'année dernière, environ un cas d'irritation cutanée sur cinq signalé dans les hôpitaux provenait de dispositifs médicaux fabriqués avec des matériaux ne répondant pas aux exigences de la norme ISO 10993-10. Les entreprises qui intègrent ces exigences dans leur processus de fabrication via des plans d'évaluation biologique appropriés rencontrent généralement moins de problèmes, car elles testent la manière dont différents matériaux réagissent au contact prolongé avec des fluides et tissus corporels.
Évaluation des risques de cytotoxicité, sensibilisation et irritation
L'ISO 10993 exige trois essais fondamentaux pour les matériaux des transducteurs de PIA :
- Cytotoxicité (ISO 10993-5) : Mesure la viabilité cellulaire après exposition au matériau (mort de 70 % des cellules autorisée)
- Sensibilisation (ISO 10993-10) : Évalue le potentiel de réaction allergique à l'aide de tests de maximisation sur cobaye
- Irritation (ISO 10993-10) : Évalue les risques d'inflammation localisée par des tests épicutanés
Les dispositifs ne passant aucun test font face à un taux de rappel par la FDA supérieur de 89 % par rapport aux alternatives pleinement conformes (Rapport sur les dispositifs médicaux, 2022).
Étude de cas : Échecs de sélection des matériaux entraînant une non-conformité
Un audit MDR UE de 2021 a révélé que 32 % des transducteurs IBP rejetés utilisaient des tubes en silicone ne disposant pas de la certification ISO 10993-33 pour un contact sanguin prolongé. Le rappel de 2,7 millions de dollars du fabricant résultait de :
- Supposer que les matériaux « de qualité médicale » satisfaisaient automatiquement aux normes de biocompatibilité
- Ne pas effectuer les tests de vieillissement accéléré pour les risques de migration chimique
- Documentation insuffisante des impacts de la stérilisation sur la sécurité des matériaux
Intégration des essais de biocompatibilité à la gestion des risques conformément à l'ISO 14971
La combinaison des données ISO 10993 avec le cadre de risque de l'ISO 14971 permet aux fabricants de :
- Quantifier les risques biologiques à l'aide de matrices gravité/probabilité des dangers
- Mettre en œuvre des mesures de contrôle telles que le remplacement des matériaux ou l'application de revêtements biocompatibles
- Surveiller les événements indésirables post-commercialisation via des processus conformes à l'ISO 13485
Cette double approche réduit de 41 % les modifications de conception en phase avancée par rapport aux essais de biocompatibilité réalisés isolément (référence qualité MedTech, 2023).
Autorisation de mise sur le marché par la FDA et exigences réglementaires mondiales pour les transducteurs IBP
Processus d'autorisation 510(k) de la FDA et équivalence substantielle
Pour les fabricants de dispositifs médicaux, obtenir une autorisation via le programme 510(k) de la FDA signifie démontrer que leur produit est sensiblement équivalent à un dispositif déjà sur le marché. Environ les trois quarts de tous les dispositifs ont été approuvés selon cette méthode en 2023, selon les données du secteur. Le respect de ces exigences nécessite des travaux importants de tests et une stricte conformité aux règles du 21 CFR Partie 820 relatives aux systèmes de qualité. Les entreprises certifiées ISO 13485 voient généralement leurs demandes traitées environ 30 % plus rapidement par la FDA. Cela semble s'expliquer par des pratiques documentaires mieux organisées, qui correspondent aux attentes des régulateurs lors de l'examen.
Problèmes d'harmonisation : différences entre le RDM UE, la FDA et d'autres marchés
Les exigences régionales divergentes créent une complexité en matière de conformité :
- Le RDM UE exige des évaluations cliniques pour l'obtention du marquage CE
- La FDA met l'accent sur l'équivalence fondée sur un prédécesseur
- L'agence japonaise PMDA exige la conformité à la norme ISO 80369-6 pour les connexions neuraxiales
- L'ANVISA brésilienne impose des tests locaux de biocompatibilité
Ces divergences obligent les fabricants à maintenir 2 à 3 variantes d'appareil, augmentant les coûts de développement de 500 000 $ à 1,2 million $ par produit (Rapport mondial sur la conformité MedTech 2024).
Tendance : Accroissement de la rigueur des exigences en matière de surveillance post-commercialisation
Les autorités de régulation exigent désormais des données sur les performances en conditions réelles, les exigences de l'FDA concernant les études post-commercialisation ayant augmenté de 40 % depuis 2021. Les fabricants doivent mettre en œuvre des systèmes électroniques de traçabilité afin de déclarer les événements indésirables dans un délai de 15 jours, soit 50 % plus rapidement qu'en 2020. Le taux d'échec aux audits de surveillance atteint désormais 22 % au niveau sectoriel, soulignant la nécessité d'une gestion proactive des risques (Revue de réglementation des dispositifs médicaux 2023).
Certifications de gestion de la qualité et de fabrication pour les fournisseurs de transducteurs IBP
Importance de l'ISO 13485 dans la conception et la production des transducteurs IBP
La certification ISO 13485 est particulièrement importante pour la gestion de la qualité parmi les fournisseurs de transducteurs IBP. Elle permet de maintenir un contrôle rigoureux à toutes les étapes, depuis la conception jusqu'à la production, et même après la mise sur le marché des produits. Ce qui distingue cette norme des standards qualité classiques, c'est sa prise en compte des exigences spécifiques aux dispositifs médicaux. Pensez, par exemple, à la traçabilité des conceptions jusqu'à leur origine, à l'assurance que la stérilisation est correctement effectuée, ou encore à la prise de décisions fondées sur une évaluation réelle des risques plutôt que sur des suppositions. L'analyse des données de 2023 concernant les rappels de dispositifs médicaux révèle également un point intéressant : les entreprises certifiées ISO 13485 ont eu environ deux fois moins de problèmes de conformité que celles qui ne l'étaient pas. En ce qui concerne les transducteurs IBP plus précisément, le respect de ces directives signifie que les capteurs de pression et les circuits fluidiques restent toujours dans des marges de sécurité strictes et fonctionnent de manière fiable tout au long de leur cycle de vie dans les hôpitaux et les cliniques.
Conformité aux bonnes pratiques de fabrication (BPF) dans la fabrication sous contrat
Le respect des bonnes pratiques de fabrication (BPF) aide les fabricants sous contrat à maintenir une cohérence dans la production de transducteurs IBP conformes aux normes FDA et MDR UE. Pourquoi les BPF sont-elles si importantes ? Elles couvrent trois domaines principaux : la tenue correcte des dossiers, le maintien de l'étalonnage des équipements et une formation adéquate du personnel. Ces aspects influencent directement la précision des mesures fournies par ces moniteurs de pression artérielle invasive. Selon les données d'audits récents, les installations pleinement conformes aux BPF obtiennent environ 98 % de réussite lors de leur premier essai de stérilité, bien au-dessus des environ 72 % observés dans les établissements ne suivant que partiellement ces règles. Lorsque les entreprises externalisent certaines étapes du processus de fabrication, comme la création des diaphragmes des transducteurs ou leur assemblage, le respect strict des BPF devient encore plus essentiel. De petites erreurs dans les procédures en salle propre chez ces prestataires peuvent gravement compromettre la qualité du produit final.
Frequently Asked Questions (FAQ)
Quel est exactement le rôle d'un transducteur IBP ?
Les transducteurs IBP convertissent les mesures de pression artérielle en signaux électriques, permettant une surveillance en temps réel de la pression artérielle lors de procédures médicales critiques.
Pourquoi la certification CE est-elle importante pour les transducteurs IBP ?
La certification CE garantit que les transducteurs IBP répondent aux normes strictes de sécurité requises pour les dispositifs médicaux vendus en Europe, assurant ainsi la sécurité des patients.
Comment les normes ISO 10993 aident-elles dans la fabrication des transducteurs IBP ?
Les normes ISO 10993 permettent aux fabricants d'évaluer la biocompatibilité des matériaux, minimisant ainsi les réactions biologiques indésirables au contact du patient.
Quel rôle jouent les organismes notifiés dans la conformité CE ?
Les organismes notifiés agissent comme des auditeurs indépendants, vérifiant que les transducteurs IBP respectent les exigences réglementaires avant leur mise sur le marché.
Pourquoi la norme ISO 13485 est-elle importante pour la gestion de la qualité dans la production de transducteurs IBP ?
L'ISO 13485 garantit que les fournisseurs de dispositifs médicaux maintiennent des normes élevées dans la conception, la production et les pratiques post-commercialisation afin d'assurer la qualité et la sécurité des produits.
Table des Matières
- Comprendre les transducteurs IBP et leur importance clinique
-
Certification CE et conformité au Règlement européen sur les dispositifs médicaux (MDR) pour les transducteurs IBP
- Ce que signifie la certification CE en matière de sécurité et de performance des dispositifs médicaux
- Étapes pour obtenir le marquage CE selon le MDR de l'UE pour les transducteurs IBP
- Rôle des organismes notifiés dans la validation de la conformité CE
- Auto-certification contre évaluation par un tiers pour les dispositifs de classe IIa
-
Normes de biocompatibilité ISO 10993 et norme de gestion des risques ISO 14971
- Pourquoi la norme ISO 10993 est-elle importante pour les composants en contact avec le patient dans les systèmes IBP
- Évaluation des risques de cytotoxicité, sensibilisation et irritation
- Étude de cas : Échecs de sélection des matériaux entraînant une non-conformité
- Intégration des essais de biocompatibilité à la gestion des risques conformément à l'ISO 14971
- Autorisation de mise sur le marché par la FDA et exigences réglementaires mondiales pour les transducteurs IBP
- Certifications de gestion de la qualité et de fabrication pour les fournisseurs de transducteurs IBP
-
Frequently Asked Questions (FAQ)
- Quel est exactement le rôle d'un transducteur IBP ?
- Pourquoi la certification CE est-elle importante pour les transducteurs IBP ?
- Comment les normes ISO 10993 aident-elles dans la fabrication des transducteurs IBP ?
- Quel rôle jouent les organismes notifiés dans la conformité CE ?
- Pourquoi la norme ISO 13485 est-elle importante pour la gestion de la qualité dans la production de transducteurs IBP ?