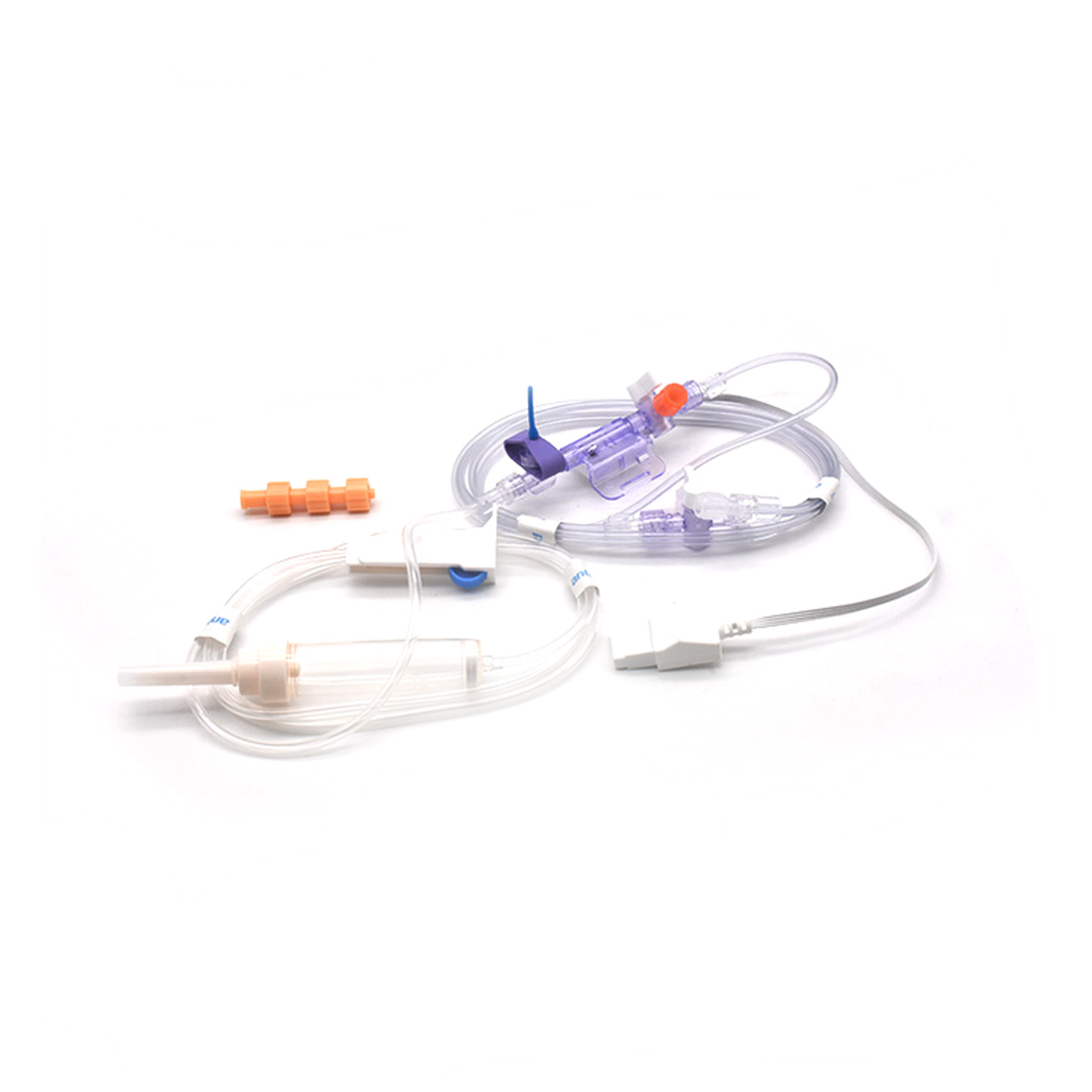
Comprensión de los transductores IBP y su importancia clínica
¿Qué es un transductor IBP y cómo funciona?
Los transductores de presión arterial invasiva (IBP) son dispositivos médicos que toman lecturas directas de la presión en las arterias a través de catéteres y las convierten en señales eléctricas para que los médicos puedan observar en tiempo real lo que está sucediendo. Estos dispositivos suelen contar con sensores estériles diseñados para detectar incluso los cambios más pequeños en los niveles de presión, y luego transmiten esta información a los monitores mediante sistemas llenos de líquido. Los modelos más recientes alcanzan una precisión de aproximadamente más o menos 2 mmHg, lo cual es fundamental para detectar caídas o aumentos peligrosos de la presión arterial durante las operaciones. Una investigación publicada el año pasado en el Journal of Critical Care Medicine reveló algo interesante: los pacientes monitoreados con estos transductores precisos tuvieron sus episodios de baja presión arterial detectados un 42 % más rápido que con técnicas no invasivas habituales. Esta clase de alerta temprana marca una gran diferencia en salas de operaciones, donde cada segundo cuenta.
Importancia clínica del monitoreo preciso de la presión arterial invasiva
Para pacientes que necesitan vasopresores o ayuda para estabilizar su circulación, la monitorización invasiva de la presión arterial (IBP) aún se considera el mejor enfoque. Los métodos oscilométricos no son suficientes cuando necesitamos lecturas de cada latido, tan importantes durante casos de shock séptico, después de accidentes graves o durante operaciones cardíacas. Los médicos dependen de la rapidez con la que reaccionan estos monitores, normalmente menos de un segundo, para tomar decisiones sobre aspectos como la administración de líquidos. La experiencia clínica ha demostrado que incluso esperar cinco minutos completos para corregir problemas de presión arterial puede llevar a una tasa de mortalidad aproximadamente un 15 % mayor. Un análisis reciente de las prácticas en unidades de cuidados intensivos reveló también algo interesante: los hospitales que implementaron configuraciones estándar de transductores IBP registraron alrededor de un 31 % menos de errores al ajustar medicamentos en sus unidades de cuidados intensivos.
Certificación CE y cumplimiento del Reglamento Europeo de Dispositivos Médicos (MDR) para transductores IBP
Qué significa la certificación CE para la seguridad y el rendimiento de los dispositivos médicos
Obtener la certificación CE de acuerdo con el Reglamento de Dispositivos Médicos de la UE (MDR) significa básicamente que un transductor IBP supera rigurosas pruebas de seguridad necesarias para vender en Europa. La marca CE indica que el producto cumple con normas importantes como la IEC 60601 para equipos electrónicos médicos y también satisface las reglas de gestión de riesgos de la ISO 14971. Para dispositivos que cumplen con el MDR, existe todo un proceso de evaluación de la compatibilidad segura de los materiales que entran en contacto con pacientes, asegurando que no causen daño celular ni irritaciones. La mayoría de los dispositivos de monitoreo de presión arterial se clasifican en la categoría Clase IIa o superior, lo que significa que los fabricantes no pueden simplemente declarar el cumplimiento por sí mismos, sino que necesitan expertos independientes que verifiquen que todo funcione correctamente antes de salir al mercado.
Pasos para obtener la marcación CE según el MDR de la UE para transductores IBP
- Clasificar el dispositivo : Determinar la clasificación de riesgo (típicamente Clase IIa para transductores IBP) según el Anexo VIII del MDR
- Implementar un sistema de gestión de calidad (QMS) : Establezca un sistema de gestión de calidad certificado según la norma ISO 13485.
- Prepare la documentación técnica : Compile las especificaciones de diseño, evaluaciones de riesgo e informes de evaluación clínica.
- Contrate un Organismo Notificado : Para dispositivos de clase IIa+, sométase a auditorías de archivos técnicos y procesos de fabricación.
- Aplique el marcado CE : Tras la evaluación de conformidad, registre el dispositivo en EUDAMED. Según SimplerQMS, el 89 % de los retrasos se deben a evaluaciones clínicas incompletas o documentación del sistema de gestión de calidad desactualizada.
Papel de los Organismos Notificados en la validación del cumplimiento del marcado CE
Los organismos notificados, o NBs por sus siglas en inglés, actúan como auditores independientes en el caso de dispositivos médicos clasificados como de mayor riesgo. Su trabajo consiste en revisar toda la documentación técnica y en acudir personalmente a los sitios de fabricación para realizar inspecciones. Al enfocarse específicamente en los transductores IBP, estos organismos verifican si todo cumple con los requisitos establecidos en el Anexo II del Reglamento de Dispositivos Médicos (MDR). Esto incluye asegurarse de que las versiones digitales cuenten con una validación adecuada del software y de que los productos desechables cumplan con estrictas normas de esterilidad. Según una encuesta reciente de 2023, la mayoría de los problemas detectados por los NBs provienen de dos áreas principales: la falta de estrategias sólidas de vigilancia postcomercialización por parte de las empresas o la insuficiencia de datos sobre biocompatibilidad. Estos hallazgos destacan los aspectos en los que los fabricantes deben centrar su atención para aprobar con éxito las auditorías.
Autocertificación frente a evaluación por terceros para dispositivos de Clase IIa
Si bien los dispositivos no estériles de Clase I pueden autodeclararse, los transductores de PAI de Clase IIa requieren supervisión obligatoria por parte de un Organismo Notificado. La autodeclaración solo se aplica a accesorios de bajo riesgo, como cables reutilizables; los componentes de detección de presión exigen una revisión completa por parte de un tercero. Las actualizaciones recientes del Reglamento de Dispositivos Médicos (MDR) exigen auditorías anuales del Organismo Notificado para los fabricantes de Clase IIa, reduciendo en un 34 % las entradas al mercado no conformes desde 2021.
Normas de biocompatibilidad ISO 10993 y de gestión de riesgos ISO 14971
Por qué es importante la ISO 10993 para los componentes en contacto con el paciente en sistemas de PAI
Para los transductores de PIA que realmente entran en contacto con pacientes, seguir las normas ISO 10993 es prácticamente obligatorio si queremos evitar esas reacciones biológicas desagradables. El objetivo principal de esta norma es determinar qué sucede cuando las personas están expuestas durante largos períodos a todo tipo de materiales, como piezas plásticas, adhesivos utilizados en el ensamblaje y diversos componentes del sensor. Según una investigación publicada el año pasado, aproximadamente uno de cada cinco casos de irritación cutánea notificados en hospitales provino de dispositivos médicos fabricados con materiales que no cumplían los requisitos de la norma ISO 10993-10. Las empresas que integran estos aspectos en sus procesos de fabricación mediante planes adecuados de evaluación biológica suelen enfrentar menos problemas, ya que analizan cómo reaccionan distintos materiales al entrar en contacto con fluidos y tejidos corporales reales a lo largo del tiempo.
Evaluación de riesgos de citotoxicidad, sensibilización e irritación
La norma ISO 10993 exige tres pruebas fundamentales para los materiales de los transductores de PIA:
- Citotoxicidad (ISO 10993-5): Mide la viabilidad celular tras la exposición al material (se permite un 70 % de muerte celular)
- Sensibilización (ISO 10993-10): Evalúa el potencial de respuesta alérgica mediante pruebas de maximación en hámster dorado
- Irritación (ISO 10993-10): Evalúa los riesgos de inflamación localizada mediante pruebas cutáneas con parches epidermales
Los dispositivos que no superan alguna prueba tienen tasas de retiro por parte de la FDA un 89 % más altas en comparación con alternativas completamente conformes (Informe de Dispositivos Médicos, 2022).
Estudio de caso: Errores en la selección de materiales que llevaron a incumplimientos
Una auditoría de la UE MDR de 2021 reveló que el 32 % de los transductores IBP rechazados utilizaban tubos de silicona sin certificación ISO 10993-33 para contacto sanguíneo prolongado. La retirada del fabricante por valor de 2,7 millones de dólares se debió a:
- Suponer que los materiales "de grado médico" cumplían automáticamente con los estándares de biocompatibilidad
- Omitir las pruebas de envejecimiento acelerado para evaluar riesgos de lixiviación química
- Documentación inadecuada de los impactos de la esterilización en la seguridad del material
Integración de las pruebas de biocompatibilidad con la gestión de riesgos según la norma ISO 14971
La combinación de los datos de la norma ISO 10993 con el marco de riesgos de la norma ISO 14971 permite a los fabricantes:
- Cuantificar los riesgos biológicos utilizando matrices de gravedad/probabilidad de peligro
- Implementar controles como sustituciones de materiales o recubrimientos biocompatibles
- Supervisar eventos adversos posteriores al mercado mediante procesos alineados con la norma ISO 13485
Este enfoque dual reduce los cambios de diseño en fases avanzadas en un 41 % en comparación con las pruebas de biocompatibilidad independientes (MedTech Quality Benchmark, 2023).
Aprobación de la FDA y requisitos regulatorios globales para transductores IBP
Proceso de aprobación 510(k) de la FDA y equivalencia sustancial
Para los fabricantes de dispositivos médicos, obtener la autorización a través del programa 510(k) de la FDA implica demostrar que su producto es sustancialmente equivalente a algo ya presente en el mercado. Aproximadamente tres cuartas partes de todos los dispositivos fueron aprobados de esta manera en 2023 según cifras de la industria. Cumplir con estos requisitos requiere un trabajo riguroso de pruebas y una estricta adherencia a las normas 21 CFR Parte 820 sobre sistemas de calidad. Las empresas que cuentan con certificación ISO 13485 tienden a ver sus solicitudes procesadas aproximadamente un 30 por ciento más rápido en la FDA. Esto parece deberse a prácticas de documentación y gestión de papeleo mejor organizadas, que coinciden con lo que los reguladores esperan ver durante la revisión.
Desafíos de armonización: Diferencias entre el Reglamento UE sobre dispositivos médicos (MDR), la FDA y otros mercados
Los requisitos regionales divergentes crean complejidad en el cumplimiento:
- El Reglamento UE sobre dispositivos médicos (MDR) exige evaluaciones clínicas para la marcación CE
- La FDA enfatiza la equivalencia basada en un producto antecedente
- La PMDA de Japón requiere cumplimiento con la norma ISO 80369-6 para conexiones neuraxiales
- ANVISA de Brasil exige pruebas locales de biocompatibilidad
Estas discrepancias obligan a los fabricantes a mantener entre 2 y 3 variantes del dispositivo, aumentando los costos de desarrollo entre 500.000 y 1,2 millones de dólares por producto (Informe Global de Cumplimiento de Tecnología Médica 2024).
Tendencia: Aumento de la exigencia en los requisitos de vigilancia postcomercialización
Actualmente, los reguladores exigen datos sobre el rendimiento en condiciones reales, con un aumento del 40 % en los requisitos de estudios postcomercialización por parte de la FDA desde 2021. Los fabricantes deben implementar sistemas electrónicos de trazabilidad para informar eventos adversos dentro de los 15 días, lo que representa una velocidad 50 % mayor que los plazos de 2020. Las tasas de falla en auditorías de vigilancia han aumentado hasta el 22 % a nivel sectorial, lo que subraya la necesidad de una gestión proactiva de riesgos (Revista de Regulación de Dispositivos Médicos 2023).
Gestión de la Calidad y Certificaciones de Fabricación para Proveedores de Transductores IBP
Importancia de la ISO 13485 en el Diseño y la Producción de Transductores IBP
La certificación ISO 13485 es realmente importante para la gestión de la calidad entre los proveedores de transductores IBP. Ayuda a mantener el control en todas las etapas, desde el diseño hasta la producción y lo que sucede después de que los productos llegan al mercado. Lo que la distingue de los estándares de calidad habituales es cómo aborda las necesidades específicas de los dispositivos médicos. Piense en aspectos como la posibilidad de rastrear los diseños hasta su origen, asegurar que la esterilización funcione adecuadamente y tomar decisiones basadas en riesgos reales en lugar de suposiciones. Analizar los datos de 2023 sobre dispositivos médicos retirados del mercado también revela algo interesante: las empresas con certificación ISO 13485 tuvieron aproximadamente la mitad de problemas para cumplir con las normas en comparación con las que no la tenían. Cuando hablamos específicamente de transductores IBP, seguir estas directrices significa que los sensores de presión y los recorridos de fluidos permanecen dentro de márgenes de seguridad estrictos y funcionan de manera confiable durante todo su ciclo de vida en hospitales y clínicas.
Alineación con la Buena Práctica de Fabricación (GMP) en la Fabricación por Contrato
Seguir las directrices de las Buenas Prácticas de Manufactura (GMP) ayuda a los fabricantes por contrato a mantener la consistencia al producir transductores de PIA que cumplan con los estándares de la FDA y el MDR de la UE. ¿Qué hace tan importante a la GMP? Bueno, abarca tres áreas principales: llevar registros adecuados, asegurar que los equipos permanezcan calibrados y capacitar debidamente al personal. Estos aspectos tienen un efecto directo sobre la precisión de las mediciones en esos monitores invasivos de presión arterial. Al analizar datos recientes de auditorías, las instalaciones plenamente conformes con la GMP obtienen alrededor de un 98 % de éxito en su primer intento durante las pruebas de esterilidad. Eso es mucho mejor que el aproximado 72 % observado en lugares que solo siguen parcialmente estas normas. Cuando las empresas subcontratan partes del proceso de fabricación, como la creación de los diafragmas del transductor o su ensamblaje, adherirse a la GMP se vuelve aún más esencial. Pequeños errores en los procedimientos de sala limpia en estos sitios tercerizados pueden afectar seriamente la calidad del producto final.
Preguntas Frecuentes (FAQ)
¿Qué hace exactamente un transductor de PIA?
Los transductores IBP convierten las lecturas de presión arterial en señales eléctricas, permitiendo el monitoreo en tiempo real de la presión arterial durante procedimientos médicos críticos.
¿Por qué es importante la certificación CE para los transductores IBP?
La certificación CE garantiza que los transductores IBP cumplan con rigurosos estándares de seguridad requeridos para dispositivos médicos vendidos en Europa, protegiendo así la seguridad del paciente.
¿Cómo ayudan las normas ISO 10993 en la fabricación de transductores IBP?
Las normas ISO 10993 permiten a los fabricantes evaluar la biocompatibilidad de los materiales, minimizando reacciones biológicas adversas por contacto con el paciente.
¿Qué papel desempeñan los Organismos Notificados en el cumplimiento de la CE?
Los Organismos Notificados actúan como auditores independientes, validando que los transductores IBP cumplan con los requisitos regulatorios antes de ingresar al mercado.
¿Por qué es importante la norma ISO 13485 para la gestión de calidad en la producción de transductores IBP?
La norma ISO 13485 asegura que los proveedores de dispositivos médicos mantengan altos estándares en el diseño, la producción y las prácticas postcomercialización para garantizar la calidad y seguridad del producto.
Tabla de Contenido
- Comprensión de los transductores IBP y su importancia clínica
-
Certificación CE y cumplimiento del Reglamento Europeo de Dispositivos Médicos (MDR) para transductores IBP
- Qué significa la certificación CE para la seguridad y el rendimiento de los dispositivos médicos
- Pasos para obtener la marcación CE según el MDR de la UE para transductores IBP
- Papel de los Organismos Notificados en la validación del cumplimiento del marcado CE
- Autocertificación frente a evaluación por terceros para dispositivos de Clase IIa
-
Normas de biocompatibilidad ISO 10993 y de gestión de riesgos ISO 14971
- Por qué es importante la ISO 10993 para los componentes en contacto con el paciente en sistemas de PAI
- Evaluación de riesgos de citotoxicidad, sensibilización e irritación
- Estudio de caso: Errores en la selección de materiales que llevaron a incumplimientos
- Integración de las pruebas de biocompatibilidad con la gestión de riesgos según la norma ISO 14971
- Aprobación de la FDA y requisitos regulatorios globales para transductores IBP
- Gestión de la Calidad y Certificaciones de Fabricación para Proveedores de Transductores IBP
-
Preguntas Frecuentes (FAQ)
- ¿Qué hace exactamente un transductor de PIA?
- ¿Por qué es importante la certificación CE para los transductores IBP?
- ¿Cómo ayudan las normas ISO 10993 en la fabricación de transductores IBP?
- ¿Qué papel desempeñan los Organismos Notificados en el cumplimiento de la CE?
- ¿Por qué es importante la norma ISO 13485 para la gestión de calidad en la producción de transductores IBP?