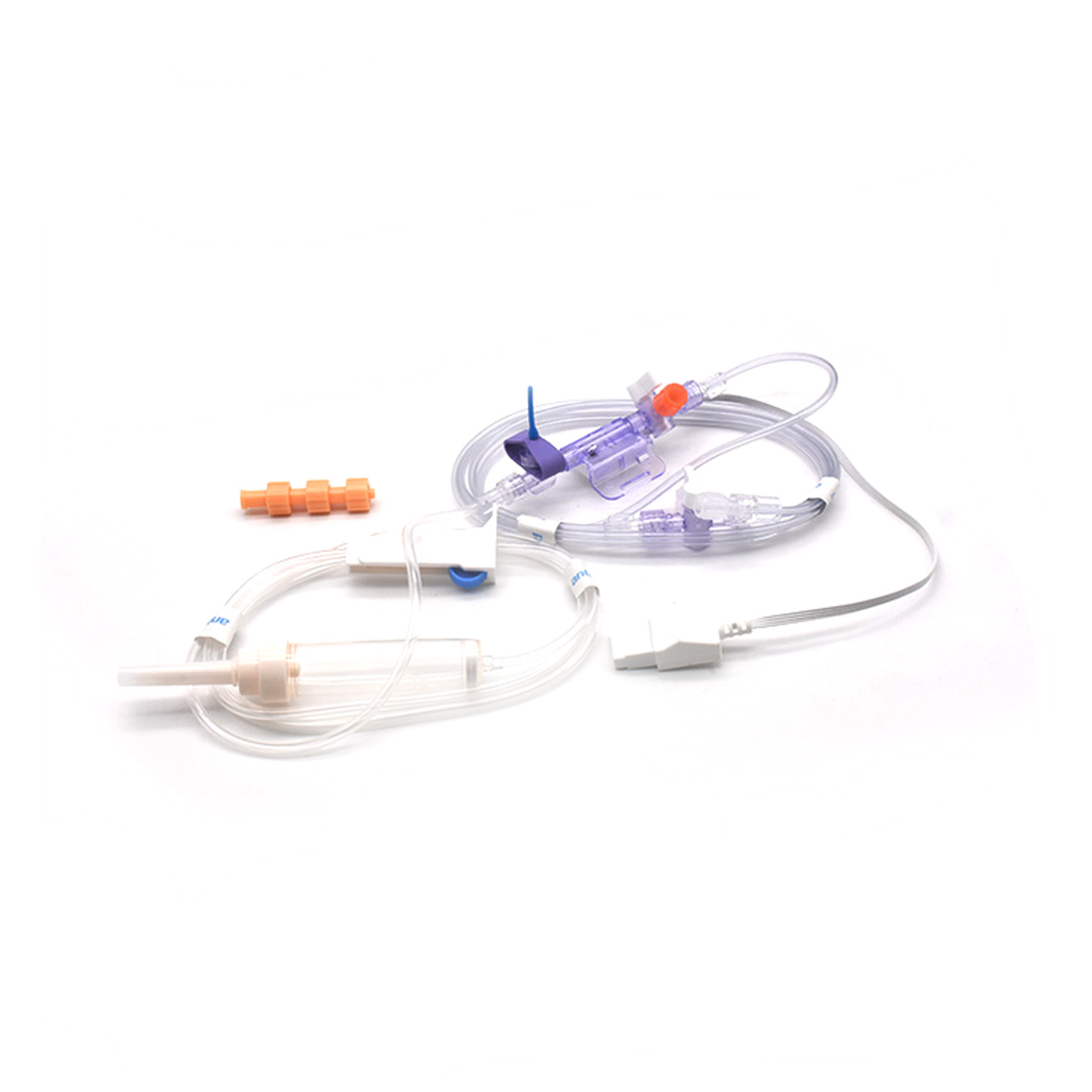
Verständnis von IBP-Transducern und ihre klinische Bedeutung
Was ist ein IBP-Transducer und wie funktioniert er?
Invasive Blutdruck-(IBP-)Wandler sind medizinische Geräte, die die tatsächlichen Druckwerte aus Arterien über Katheter erfassen und in elektrische Signale umwandeln, sodass Ärzte den Vorgang in Echtzeit verfolgen können. Diese Geräte verfügen typischerweise über sterile gehaltene Sensoren, die selbst kleinste Druckschwankungen erfassen, und leiten diese Informationen anschließend über flüssigkeitsgefüllte Systeme an die Monitore weiter. Die neueren Modelle erreichen eine Genauigkeit von etwa ±2 mmHg, was besonders wichtig ist, um gefährliche Abfälle oder Anstiege des Blutdrucks während Operationen frühzeitig zu erkennen. Eine im vergangenen Jahr im Journal of Critical Care Medicine veröffentlichte Studie zeigte darüber hinaus etwas Interessantes: Bei Patienten, die mit diesen präzisen Wandlern überwacht wurden, wurden Episoden mit niedrigem Blutdruck 42 % schneller erkannt als mit herkömmlichen nicht-invasiven Verfahren. Solche Frühwarnungen machen einen großen Unterschied in Operationssälen, wo jede Sekunde zählt.
Klinische Bedeutung einer genauen invasiven Blutdruckmessung
Für Patienten, die Vasopressoren benötigen oder bei der Stabilisierung ihrer Durchblutung Unterstützung benötigen, gilt die invasive Blutdruckmessung (IBP) weiterhin als beste Methode. Oszillometrische Verfahren sind unzureichend, wenn wir beatmäßige Messwerte benötigen, die insbesondere bei septischem Schock, nach schweren Unfällen oder während Herzoperationen von entscheidender Bedeutung sind. Ärzte verlassen sich auf die schnelle Reaktionszeit dieser Monitore, die typischerweise unter einer Sekunde liegt, um Entscheidungen über Maßnahmen wie die Gabe von Infusionsflüssigkeiten zu treffen. Klinische Erfahrungen haben gezeigt, dass bereits eine Wartezeit von fünf Minuten bei der Korrektur von Blutdruckstörungen zu einer um etwa 15 % höheren Sterblichkeitsrate führen kann. Eine aktuelle Untersuchung intensivmedizinischer Praktiken hat zudem Folgendes ergeben: Krankenhäuser, die standardisierte IBP-Sensor-Setups eingeführt hatten, wiesen in ihren Intensivstationen rund 31 % weniger Fehler bei der Medikamentendosierung auf.
CE-Zertifizierung und EU-MDR-Konformität für IBP-Sensoren
Bedeutung der CE-Zertifizierung für Sicherheit und Leistung medizinischer Geräte
Die CE-Zertifizierung gemäß der EU-Verordnung über Medizinprodukte (MDR) bedeutet im Wesentlichen, dass ein IBP-Sensor strenge Sicherheitsprüfungen bestanden hat, die zum Verkauf in Europa erforderlich sind. Die CE-Kennzeichnung zeigt an, dass das Produkt wichtigen Normen wie IEC 60601 für medizinische elektronische Geräte entspricht und zudem die Risikomanagementvorschriften nach ISO 14971 erfüllt. Bei Geräten, die den MDR-Vorgaben entsprechen, wird umfassend geprüft, wie gut Materialien, die mit Patienten in Berührung kommen, sicher zusammenwirken, um sicherzustellen, dass sie keine Zellschädigungen oder Reizungen verursachen. Die meisten Blutdruckmessgeräte fallen in die Kategorie Klasse IIa oder höher, was bedeutet, dass Hersteller die Konformität nicht einfach selbst erklären können, sondern unabhängige Experten benötigen, um sicherzustellen, dass alles ordnungsgemäß funktioniert, bevor das Gerät auf den Markt kommt.
Schritte zur Erlangung der CE-Kennzeichnung gemäß der EU-MDR für IBP-Transducer
- Gerät klassifizieren : Risikoklassifizierung bestimmen (typischerweise Klasse IIa für IBP-Transducer) gemäß Anhang VIII der MDR.
- Ein QMS implementieren : Ein ISO-13485-zertifiziertes Qualitätsmanagementsystem einrichten.
- Technische Dokumentation erstellen : Konstruktionsvorgaben, Risikobewertungen und klinische Bewertungsberichte zusammenstellen.
- Eine benannte Stelle hinzuziehen : Für Geräte der Klasse IIa+ Prüfungen der technischen Unterlagen und Produktionsverfahren durchführen lassen.
- CE-Kennzeichnung anbringen : Nach der Konformitätsbewertung das Gerät in EUDAMED registrieren. Laut SimplerQMS gehen 89 Prozent der Verzögerungen auf unvollständige klinische Bewertungen oder veraltete QMS-Dokumentation zurück.
Rolle benannter Stellen bei der Validierung der CE-Konformität
Die benannten Stellen, kurz NBs, fungieren als unabhängige Prüfer bei medizinischen Geräten, die als höheres Risiko eingestuft werden. Ihre Aufgabe umfasst die Durchsicht aller technischen Unterlagen sowie tatsächliche Besuche an Fertigungsstandorten zur Inspektion. Bei der Betrachtung von IBP-Transducern prüfen diese Stellen, ob alles mit den Anforderungen gemäß Anhang II der MDR übereinstimmt. Dazu gehört, dass digitale Versionen eine ordnungsgemäße Softwarevalidierung aufweisen und Einmaldisposables strengen Sterilitätsanforderungen genügen. Laut einer aktuellen Umfrage aus dem Jahr 2023 resultieren die meisten von den NBs festgestellten Probleme aus zwei Hauptbereichen: Unternehmen verfügen nicht über fundierte Strategien zur Nachmarktüberwachung oder liefern nicht ausreichende Daten zur Biokompatibilität. Diese Erkenntnisse verdeutlichen, worauf sich Hersteller konzentrieren müssen, um Audits erfolgreich zu bestehen.
Selbstzertifizierung vs. Drittprüfung für Geräte der Klasse IIa
Während nicht sterile Geräte der Klasse I eine Selbstzertifizierung vornehmen können, erfordern Druckwandler der Klasse IIa für invasiven Blutdruck (IBP) die obligatorische Überwachung durch eine benannte Stelle. Die Selbstzertifizierung gilt nur für niedrigriskante Zubehörteile wie wiederverwendbare Kabel; drucksensorische Komponenten erfordern eine vollständige Prüfung durch eine unabhängige Drittpartei. Aktuelle MDR-Updates schreiben jährliche Audits durch die benannte Stelle für Hersteller von Klasse-IIa-Geräten vor und haben seit 2021 die Zahl nicht konformer Markteintritte um 34 % reduziert.
Biokompatibilität nach ISO 10993 und Risikomanagement nach ISO 14971
Warum ISO 10993 für patientenberührende Komponenten in IBP-Systemen wichtig ist
Für IBP-Sensoren, die tatsächlich Patienten berühren, ist die Einhaltung der ISO 10993-Standards nahezu zwingend erforderlich, wenn wir jene unangenehmen biologischen Reaktionen vermeiden wollen. Der gesamte Sinn dieses Standards besteht darin, herauszufinden, was passiert, wenn Menschen über längere Zeiträume diversen Materialien ausgesetzt sind, wie beispielsweise Kunststoffteilen, Klebstoffen, die bei der Montage verwendet werden, und verschiedenen Sensorkomponenten. Laut einer im vergangenen Jahr veröffentlichten Studie entfiel etwa jeder fünfte im Krankenhaus gemeldete Fall von Hautreizungen auf medizinische Geräte, die aus Materialien hergestellt wurden, welche die Anforderungen nach ISO 10993-10 nicht erfüllten. Unternehmen, die diese Aspekte mithilfe geeigneter Biologischer Evaluierungspläne in ihren Herstellungsprozess integrieren, haben tendenziell weniger Probleme, da sie testen, wie verschiedene Materialien reagieren, wenn sie über einen längeren Zeitraum mit echten Körperflüssigkeiten und Geweben in Kontakt kommen.
Bewertung von Zytotoxizität, Sensibilisierung und Reizungsrisiken
ISO 10993 schreibt drei Kernprüfungen für Materialien von IBP-Transducern vor:
- Zytotoxizität (ISO 10993-5): Misst die Zellvitalität nach Materialexposition (’70 % Zelltod erlaubt)
- Sensibilisierung (ISO 10993-10): Bewertet das Potenzial für allergische Reaktionen mittels Guinea-Pig-Maximierungstests
- Reizung (ISO 10993-10): Beurteilt das Risiko lokaler Entzündungen durch epidermale Pflastertests
Geräte, die bei einem Test versagen, weisen eine 89 % höhere FDA-Rückrufquote auf als vollständig konforme Alternativen (Medical Device Reporting, 2022).
Fallstudie: Fehlerhafte Materialauswahl führt zu Nichteinhaltung
Eine EU MDR-Auditierung aus dem Jahr 2021 ergab, dass 32 % der abgelehnten IBP-Transducer Silikonschläuche verwendeten, die nicht über die ISO 10993-33-Zertifizierung für längeren Blutkontakt verfügten. Der Hersteller-Rückruf in Höhe von 2,7 Mio. USD resultierte aus:
- Annahme, dass „medizinisches Material“ automatisch die Biokompatibilitätsnormen erfüllt
- Unterlassung beschleunigter Alterungstests hinsichtlich des Auslaufens von Chemikalien
- Unzureichende Dokumentation der Auswirkungen der Sterilisation auf die Materialsicherheit
Integration der Biokompatibilitätsprüfung in das Risikomanagement gemäß ISO 14971
Die Kombination von ISO 10993-Daten mit dem Risikorahmenwerk der ISO 14971 ermöglicht es Herstellern,
- Biologische Risiken mithilfe von Gefahrenschwere/-wahrscheinlichkeits-Matrizen zu quantifizieren
- Maßnahmen wie Materialersetzungen oder biokompatible Beschichtungen umzusetzen
- Nachmarktliche unerwünschte Ereignisse durch ISO 13485-konforme Prozesse zu überwachen
Dieser zweigleisige Ansatz reduziert späte Konstruktionsänderungen um 41 % im Vergleich zur alleinigen Biokompatibilitätsprüfung (MedTech Quality Benchmark, 2023).
FDA-Zulassung und globale regulatorische Anforderungen für IBP-Sensoren
FDA 510(k)-Zulassungsverfahren und Nachweis der wesentlichen Gleichwertigkeit
Für Hersteller medizinischer Geräte bedeutet die Zulassung über das FDA-Programm 510(k), nachzuweisen, dass ihr Produkt im Wesentlichen gleichwertig zu einem bereits auf dem Markt befindlichen Produkt ist. Laut Branchenangaben wurden etwa drei Viertel aller Geräte im Jahr 2023 auf diese Weise zugelassen. Die Erfüllung dieser Anforderungen erfordert umfangreiche Testarbeiten und strikte Einhaltung der Vorschriften des 21 CFR Part 820 bezüglich Qualitätsmanagementsystemen. Unternehmen mit ISO-13485-Zertifizierung sehen ihre Anträge bei der FDA tendenziell etwa 30 Prozent schneller bearbeitet. Dies scheint auf besser organisierte Unterlagen und Dokumentationspraktiken zurückzuführen zu sein, die den Erwartungen der Aufsichtsbehörden während der Prüfung entsprechen.
Herausforderungen bei der Harmonisierung: Unterschiede zwischen EU-MDR, FDA und anderen Märkten
Voneinander abweichende regionale Anforderungen schaffen Compliance-Komplexität:
- Die EU-MDR schreibt klinische Bewertungen für die CE-Kennzeichnung vor
- Die FDA legt den Schwerpunkt auf die Äquivalenz zu einem Vorläuferprodukt (predicate-based equivalence)
- Japans PMDA verlangt die Konformität mit ISO 80369-6 für neuraxiale Anschlüsse
- Brasiliens ANVISA setzt lokale Biokompatibilitätsprüfungen durch
Diese Diskrepanzen zwingen Hersteller, 2–3 Gerätevarianten aufrechtzuerhalten, was die Entwicklungskosten pro Produkt um 500.000–1,2 Mio. USD erhöht (Globaler MedTech Compliance Report 2024).
Trend: Zunehmende Verschärfung der Anforderungen an das Nachmarktaufsichtsverfahren
Regulierungsbehörden verlangen nun Daten zur Leistung im praktischen Einsatz; die Anforderungen der FDA für Nachmarktstudien sind seit 2021 um 40 % gestiegen. Hersteller müssen elektronische Rückverfolgbarkeitssysteme implementieren, um Vorfälle innerhalb von 15 Tagen zu melden – 50 % schneller als die Fristen aus dem Jahr 2020. Die Durchfallquoten bei Überwachungsaudits sind branchenweit auf 22 % angestiegen, was die Notwendigkeit eines proaktiven Risikomanagements unterstreicht (Medical Device Regulatory Journal 2023).
Qualitätsmanagement und Fertigungszertifizierungen für IBP-Transducer-Lieferanten
Bedeutung von ISO 13485 bei der Konstruktion und Produktion von IBP-Transducern
Die ISO-13485-Zertifizierung ist für das Qualitätsmanagement bei IBP-Transducer-Lieferanten von großer Bedeutung. Sie trägt dazu bei, die Kontrolle über alle Phasen – von der Entwicklung über die Produktion bis hin zur Zeit nach der Markteinführung der Produkte – aufrechtzuerhalten. Im Unterschied zu allgemeinen Qualitätsstandards berücksichtigt sie gezielt die spezifischen Anforderungen medizinischer Geräte. Dazu gehören beispielsweise die Rückverfolgbarkeit von Entwicklungen, die Gewährleistung einer wirksamen Sterilisation und risikobasierte Entscheidungsfindung statt Annahmen. Daten aus dem Jahr 2023 zu zurückgerufenen medizinischen Geräten zeigen zudem ein interessantes Ergebnis: Unternehmen mit ISO-13485-Zertifizierung wiesen etwa halb so viele Mängel bei der Einhaltung von Vorschriften auf wie solche ohne Zertifizierung. Bei IBP-Transducern bedeutet die Einhaltung dieser Richtlinien konkret, dass Drucksensoren und Fluidwege innerhalb strenger Sicherheitsgrenzen bleiben und im gesamten Lebenszyklus in Krankenhäusern und Arztpraxen zuverlässig funktionieren.
Übereinstimmung mit den Grundsätzen der Guten Herstellungspraxis (GMP) bei der Auftragsfertigung
Die Einhaltung der Richtlinien für Gute Herstellungspraxis (GMP) hilft Auftragsherstellern, bei der Produktion von IBP-Transducern sowohl die FDA- als auch die EU-MDR-Normen einzuhalten und gleichzeitig die Konsistenz sicherzustellen. Warum ist GMP so wichtig? Nun, es umfasst drei Hauptbereiche: ordnungsgemäße Dokumentation, Sicherstellung einer korrekten Kalibrierung der Geräte und eine angemessene Schulung des Personals. Diese Aspekte wirken sich direkt auf die Genauigkeit der Messergebnisse bei invasiven Blutdruckmonitoren aus. Aktuelle Audit-Daten zeigen, dass Produktionsstätten, die vollständig GMP-konform arbeiten, bei Sterilitätstests etwa 98 % Erfolgsquote beim ersten Versuch erzielen. Das liegt deutlich über den rund 72 %, die an Standorten erreicht werden, die diese Vorschriften nur teilweise befolgen. Wenn Unternehmen Teile des Herstellungsprozesses, wie die Herstellung der Transducer-Diaphragmen oder deren Montage, auslagern, wird die Einhaltung von GMP noch wichtiger. Kleine Fehler in den Reinraumverfahren bei diesen externen Partnern können die Qualität des Endprodukts erheblich beeinträchtigen.
Frequently Asked Questions (FAQ)
Was genau macht ein IBP-Transducer?
IBP-Transducer wandeln Druckmessungen aus Arterien in elektrische Signale um, wodurch eine Echtzeitüberwachung des Blutdrucks während kritischer medizinischer Eingriffe ermöglicht wird.
Warum ist die CE-Zertifizierung für IBP-Transducer wichtig?
Die CE-Zertifizierung stellt sicher, dass IBP-Transducer strenge Sicherheitsstandards erfüllen, die für medizinische Geräte, die in Europa verkauft werden, erforderlich sind, und schützt so die Patientensicherheit.
Wie helfen die ISO-10993-Normen bei der Herstellung von IBP-Transducern?
Die ISO-10993-Normen ermöglichen es Herstellern, Materialien auf Biokompatibilität zu prüfen, um unerwünschte biologische Reaktionen durch Patientenkontakt zu minimieren.
Welche Rolle spielen benannte Stellen bei der CE-Konformität?
Benannte Stellen fungieren als unabhängige Prüfer und bestätigen, dass IBP-Transducer die regulatorischen Anforderungen erfüllen, bevor sie auf den Markt gelangen.
Warum ist ISO 13485 wichtig für das Qualitätsmanagement bei der Produktion von IBP-Transducern?
ISO 13485 stellt sicher, dass Lieferanten medizinischer Geräte hohe Standards in den Bereichen Entwicklung, Produktion und Nachmarktpraktiken einhalten, um Produktqualität und -sicherheit zu gewährleisten.
Inhaltsverzeichnis
- Verständnis von IBP-Transducern und ihre klinische Bedeutung
- CE-Zertifizierung und EU-MDR-Konformität für IBP-Sensoren
- Biokompatibilität nach ISO 10993 und Risikomanagement nach ISO 14971
- FDA-Zulassung und globale regulatorische Anforderungen für IBP-Sensoren
- Qualitätsmanagement und Fertigungszertifizierungen für IBP-Transducer-Lieferanten
-
Frequently Asked Questions (FAQ)
- Was genau macht ein IBP-Transducer?
- Warum ist die CE-Zertifizierung für IBP-Transducer wichtig?
- Wie helfen die ISO-10993-Normen bei der Herstellung von IBP-Transducern?
- Welche Rolle spielen benannte Stellen bei der CE-Konformität?
- Warum ist ISO 13485 wichtig für das Qualitätsmanagement bei der Produktion von IBP-Transducern?