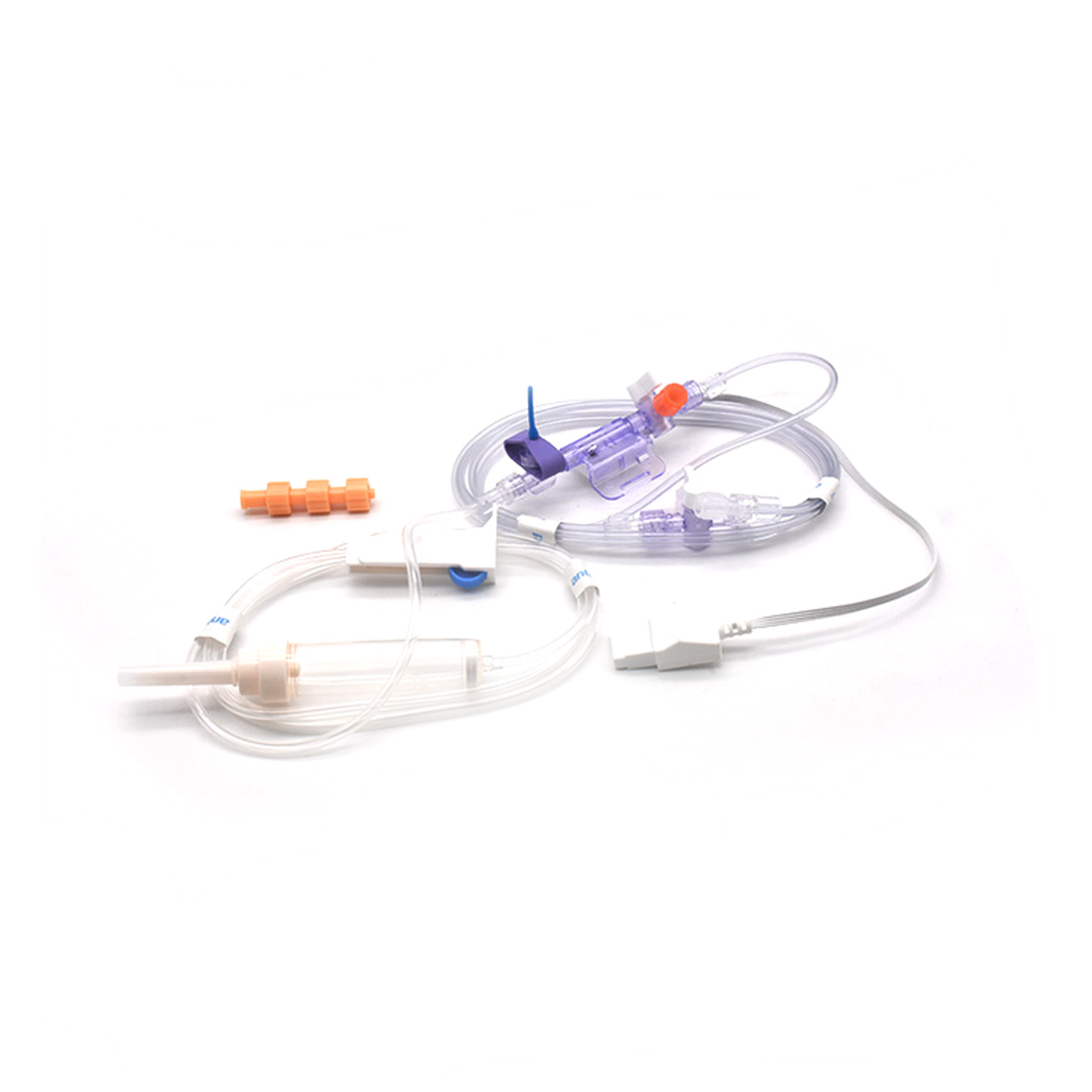
Разбиране на IBP трансдюсерите и тяхното клинично значение
Какво е IBP преобразувател и как работи?
Трансдюсерите за инвазивно измерване на кръвното налягане (IBP) са медицински устройства, които извършват реални измервания на налягането в артериите чрез свързване с катетри и го преобразуват в електрически сигнали, така че лекарите да могат да наблюдават процеса в реално време. Тези устройства обикновено разполагат със стерилни сензори, които улавят дори най-малките промени в нивата на налягане и след това предават цялата информация към мониторите чрез флуидни системи. Новите модели постигат точност от около плюс или минус 2 mmHg, което има голямо значение при откриването на опасни спадове или скокове в кръвното налягане по време на операции. Проучване, публикувано миналата година в списание Journal of Critical Care Medicine, показа още нещо интересно – при пациенти, наблюдавани с тези прецизни трансдюсери, епизодите с ниско кръвно налягане са били засечени с 42% по-бързо в сравнение с обикновените неинвазивни методи. Такова ранно предупреждение прави голяма разлика в операционните зали, където всеки секунда има значение.
Клинично значение на точното инвазивно наблюдение на кръвното налягане
За пациенти, нуждаещи се от вазопресори или помощ при стабилизиране на кръвообращението, инвазивното измерване на кръвното налягане (IBP) все още се счита за най-добрия подход. Осцилометричните методи просто не са достатъчни, когато се нуждаем от данни за всеки отделен удар, които имат голямо значение при случаи на септичен шок, след тежки злополуки или по време на сърдечни операции. Лекарите разчитат на бързата реакция на тези монитори, обикновено под една секунда, за да вземат решения относно например прилагането на течности. От клиническия опит знаем, че дори изчакването на цели пет минути за коригиране на проблеми с кръвното налягане може да доведе до около 15% по-висока смъртност. Наскорошно проучване на практиките в интензивни отделения показа още нещо интересно: болниците, които приложиха стандартни IBP преобразуватели, отбелязаха около 31% по-малко грешки при коригиране на медикаментите в своите интензивни отделения.
Сертифициране по СЕ и съответствие с Регламент за медицински изделия на ЕС (EU MDR) за IBP преобразуватели
Какво означава сертифицирането по СЕ за безопасността и производителността на медицинските устройства
Получаването на CE сертификат съгласно Регламента за медицински изделия на ЕС (MDR) по същество означава, че IBP трансдюсерът е издържал строги проверки за безопасност, необходими за продажба в Европа. Означението CE показва, че продуктът отговаря на важни стандарти като IEC 60601 за медицинско електронно оборудване и също така изпълнява изискванията за управление на риска от ISO 14971. За устройствата, които спазват разпоредбите на MDR, съществува цял процес на тестване на съвместимостта на материалите, които докосват пациенти, с цел осигуряване, че няма да причинят увреждане на клетките или раздразнения. Повечето устройства за мониторинг на кръвното налягане попадат в категория IIa или по-висока, което означава, че производителите не могат просто да декларират съответствие сами, а се нуждаят от независими експерти, които да потвърдят, че всичко работи правилно, преди устройствата да бъдат пуснати на пазара.
Стъпки за постигане на CE означение по силата на MDR на ЕС за IBP трансдюсери
- Класифициране на устройството : Определяне на класификацията по рискове (обикновено клас IIa за IBP трансдюсери) според Приложение VIII на MDR.
- Въвеждане на система за управление на качеството (QMS) : Създаване на система за управление на качеството, съответстваща на ISO 13485.
- Подготвяне на техническа документация : Съставяне на спецификации за конструкцията, оценки на риска и доклади за клинична оценка.
- Включване на уведомено тяло : За устройства от клас IIa и по-горе, провеждане на одити на техническата документация и производствените процеси.
- Поставяне на CE маркировка : След оценка на съответствието, регистриране на устройството в EUDAMED. Според SimplerQMS, 89% от закъсненията се дължат на непълни клинични оценки или остаряла документация по системата за управление на качеството.
Ролята на уведомените тела при валидиране на съответствието с CE изискванията
Нотифицираните органи, или НБ за кратко, служат като независими одитори, когато става въпрос за медицински изделия, класифицирани като с по-висок риск. Работата им включва да преглеждат всички технически документи и да се явят на производствените обекти за проверки. Когато се разглеждат специално IBP преобразувателите, тези органи проверяват дали всичко съответства на изискванията, изложени в приложение II към MDR. Това включва да се уверите, че цифровите версии имат подходящо потвърждение на софтуера и че еднократните продукти отговарят на строги стандарти за стерилност. Според скорошно проучване от 2023 г. повечето от проблемите, открити от НБ, произтичат от две основни области: компаниите нямат солидни стратегии за надзор след пускането на пазара или не предоставят достатъчно данни за биосъвместимост. Тези констатации подчертават къде производителите трябва да съсредоточат вниманието си, ако искат да преминат успешно одитите.
Самосертифициране срещу оценка от трета страна за изделия от клас IIa
Докато клас I нестерилни устройства могат да се самосертифицират, трансдюсерите за ИАД от клас IIa изискват задължителен надзор от уведомена организация. Самосертифицирането се прилага само за аксесоари с нисък риск, като многократно използваеми кабели; компонентите за измерване на налягане изискват пълна проверка от трета страна. Скоростните актуализации на Регламента за медицински изделия (MDR) предвиждат годишни одити от уведомена организация за производителите на устройства от клас IIa, което намалява несъответстващите влизания на пазара с 34% от 2021 г. насам.
ISO 10993 Стандарти за биосъвместимост и ISO 14971 Управление на риска
Защо стандартът ISO 10993 е важен за компонентите, контактуващи с пациент, в системите за ИАД
За трансдюсери за IBP, които всъщност докосват пациенти, следването на стандарта ISO 10993 е почти задължително, ако искаме да избегнем неприятните биологични реакции. Цялата цел на този стандарт е да се установи какво се случва, когато хората бъдат изложени в продължение на дълги периоди на различни вещества като пластмасови части, лепила, използвани при сглобяването, и различни сензорни компоненти. Според проучване, публикувано миналата година, около един от всеки пет случая на раздразнения на кожата, регистрирани в болници, са причинени от медицински устройства, направени от материали, които не отговарят на изискванията на ISO 10993-10. Компаниите, които включват тези изисквания в производствения си процес чрез подходящи планове за биологична оценка, обикновено срещат по-малко проблеми, тъй като тестват как различните материали реагират при контакт с истински телесни течности и тъкани в продължение на време.
Оценка на рисковете от цитотоксичност, сенсибилизация и раздразнения
ISO 10993 изисква три основни теста за материалите на трансдюсерите за IBP:
- Цитотоксичност (ISO 10993-5): Измерва жизнеността на клетките след въздействие на материала (допуска се до 70% смъртност на клетките)
- Сенсибилизация (ISO 10993-10): Оценява потенциала за алергична реакция чрез тестове с максимална сенсибилизация при морски свинчета
- Иритация (ISO 10993-10): Оценява риска от локализирано възпаление чрез епидермални тестове с пластири
Устройствата, които не изпълняват изискванията по който и да е тест, имат с 89% по-висок шанс за отзиване от FDA в сравнение с напълно съвместимите алтернативи (Докладване за медицински изделия, 2022 г.).
Примерно проучване: Грешки при избора на материали, довели до несъответствие
Аудит по EU MDR през 2021 г. разкри, че 32% от отхвърлените IBP трансдюсери използват силиконова тръба, непреминаваща сертифициране по ISO 10993-33 за продължителен контакт с кръв. Отзиването на производителя за 2,7 млн. долара се дължи на:
- Предположението, че материалите от "медицински клас" автоматично отговарят на изискванията за биосъвместимост
- Пропускане на ускорени тестове за стареене относно рисковете от изтичане на химикали
- Недостатъчна документация за въздействието на стерилизацията върху безопасността на материалите
Интегриране на изпитвания за биосъвместимост с управлението на риска според ISO 14971
Комбинирането на данните от ISO 10993 с рамката за риск от ISO 14971 позволява на производителите да:
- Количествено определят биологичните рискове, използвайки матрици за тежест/вероятност на опасности
- Въвеждат контролни мерки като замяна на материали или биосъвместими покрития
- Проследяват нежелани събития след пускане на пазара чрез процеси, съобразени с ISO 13485
Този двоен подход намалява промените в дизайна на късен етап с 41% в сравнение с изолираното тестване за биосъвместимост (MedTech Quality Benchmark, 2023).
Одобряване от FDA и глобални регулаторни изисквания за IBP трансдюсери
Процес на одобрение FDA 510(k) и съществено сходство
За производителите на медицински устройства, получаването на разрешение чрез програмата на FDA 510(k) означава да докажат, че техният продукт е съществено еквивалентен на вече наличен на пазара. Според данни от индустрията около три четвърти от всички устройства са били одобрени по този начин през 2023 г. Изпълнението на тези изисквания изисква сериозна изпитвателна дейност и стриктно спазване на правилата от 21 CFR Part 820 относно системите за качество. Компаниите със сертифициране по ISO 13485 обикновено виждат своите заявки обработвани приблизително с 30 процента по-бързо от страна на FDA. Това изглежда се дължи на по-добре организирана документация и практики при подаване на документи, които отговарят на това, което регулаторите очакват да видят по време на прегледа.
Предизвикателства при хармонизация: Разлики между EU MDR, FDA и други пазари
Различните регионални изисквания създават сложност при спазването на изискванията:
- EU MDR изисква клинични оценки за получаване на CE маркировка
- FDA набляга на еквивалентност, базирана на предшестващ модел
- Японската PMDA изисква съответствие с ISO 80369-6 за неврални свързвания
- Бразилската ANVISA прилага местни изследвания за биосъвместимост
Тези несъответствия принуждават производителите да поддържат 2–3 варианта на устройствата, което увеличава разходите за разработка с 500 хил. до 1,2 млн. долара за продукт (Доклад за съответствие в глобалния медтехник 2024).
Тенденция: Увеличаване на строгостта на изискванията за наблюдение след пускане на пазара
Регулаторните органи вече изискват данни за реална ефективност, като изискванията на FDA за проучвания след пускане на пазара са нараснали с 40% от 2021 г. Производителите трябва да внедрят електронни системи за проследяване, за да докладват нежелани събития в рамките на 15 дни – с 50% по-бързо в сравнение с графиките от 2020 г. Стойностите на неуспехите при одити за наблюдение са нараснали до 22% в целия сектор, което подчертава необходимостта от превантивно управление на риска (Списание за регулаторика на медицински изделия 2023).
Сертификати за управление на качеството и производство за доставчици на IBP трансдюсери
Значението на ISO 13485 при проектирането и производството на IBP трансдюсери
Сертификатът ISO 13485 има голямо значение за управлението на качеството сред доставчиците на IBP трансдюсери. Той помага за поддържане на контрол на всички етапи – от проектирането през производството до действията след излизането на продуктите на пазара. Онова, което отличава този стандарт от обикновените изисквания за качество, е начина, по който се справя с конкретните нужди на медицинските устройства. Помислете за неща като възможността за проследяване на проекти до техния произход, осигуряване на правилната стерилизация и вземане на решения въз основа на реални рискове, а не на предположения. Анализът на данни от 2023 г. относно оттеглени медицински устройства показва още нещо интересно: компании с ISO 13485 сертификация са имали около два пъти по-малко проблеми със спазването на стандарти в сравнение с тези без такава. Когато говорим конкретно за IBP трансдюсери, следването на тези насоки означава, че сензорите за налягане и флуидните пътища ще остават в строгите граници на безопасността и ще работят надеждно през целия си жизнен цикъл в болници и клиники.
Съответствие с добрата производствена практика (GMP) при договорно производство
Следването на насоките за добра производствена практика (GMP) помага на договорните производители да поддържат последователност при изграждането на IBP трансдюсери, отговарящи на изискванията на FDA и EU MDR. Защо GMP е толкова важно? Ами, то обхваща три основни области: водене на правилна документация, осигуряване на калибриране на оборудването и правилно обучение на персонала. Тези аспекти имат пряко влияние върху точността на измерванията за тези инвазивни монитори за кръвно налягане. Според данни от последните одити, обектите, напълно съответстващи на GMP, постигат около 98% успех при първия опит в тестовете за стерилност. Това е значително по-добре от приблизително 72%, регистрирани при места, които следват частично тези правила. Когато компании извъншят части от производствения процес, като създаването на мембраните на трансдюсера или тяхната сглобка, спазването на GMP става още по-съществено. Малки грешки в процедурите в чисти стаи при тези странични доставчици могат сериозно да повлияят върху крайното качество на продукта.
Често задавани въпроси (FAQ)
Какво точно прави един IBP трансдюсер?
IBP трансдюсерите преобразуват показанията за налягане от артериите в електрически сигнали, което позволява непрекъснат мониторинг на кръвното налягане по време на критични медицински процедури.
Защо е важно CE сертифицирането за IBP трансдюсери?
CE сертифицирането гарантира, че IBP трансдюсерите отговарят на строгите изисквания за безопасност, необходими за медицинските устройства, продавани в Европа, и осигурява безопасността на пациентите.
Как ISO 10993 стандарти помагат при производството на IBP трансдюсери?
Стандартите ISO 10993 позволяват на производителите да оценяват материали за биосъвместимост, като по този начин се минимизират нежелани биологични реакции при контакт с пациента.
Каква роля играят уведомените органи при спазването на CE изискванията?
Уведомените органи действат като независими одитори, които потвърждават, че IBP трансдюсерите отговарят на регулаторните изисквания преди влизане на пазара.
Защо ISO 13485 е важен за управлението на качеството при производството на IBP трансдюсери?
ISO 13485 гарантира, че доставчиците на медицински устройства поддържат високи стандарти в проектирането, производството и дейностите след пускане на пазара, за да осигурят качеството и безопасността на продуктите.
Съдържание
- Разбиране на IBP трансдюсерите и тяхното клинично значение
-
Сертифициране по СЕ и съответствие с Регламент за медицински изделия на ЕС (EU MDR) за IBP преобразуватели
- Какво означава сертифицирането по СЕ за безопасността и производителността на медицинските устройства
- Стъпки за постигане на CE означение по силата на MDR на ЕС за IBP трансдюсери
- Ролята на уведомените тела при валидиране на съответствието с CE изискванията
- Самосертифициране срещу оценка от трета страна за изделия от клас IIa
-
ISO 10993 Стандарти за биосъвместимост и ISO 14971 Управление на риска
- Защо стандартът ISO 10993 е важен за компонентите, контактуващи с пациент, в системите за ИАД
- Оценка на рисковете от цитотоксичност, сенсибилизация и раздразнения
- Примерно проучване: Грешки при избора на материали, довели до несъответствие
- Интегриране на изпитвания за биосъвместимост с управлението на риска според ISO 14971
- Одобряване от FDA и глобални регулаторни изисквания за IBP трансдюсери
- Сертификати за управление на качеството и производство за доставчици на IBP трансдюсери
-
Често задавани въпроси (FAQ)
- Какво точно прави един IBP трансдюсер?
- Защо е важно CE сертифицирането за IBP трансдюсери?
- Как ISO 10993 стандарти помагат при производството на IBP трансдюсери?
- Каква роля играят уведомените органи при спазването на CE изискванията?
- Защо ISO 13485 е важен за управлението на качеството при производството на IBP трансдюсери?